
Directives sur la façon de remplir le formulaire de présentation pour les médicaments
Mise à jour : le 31 mars 2021
Directive
En ce qui concerne les demandes d'identification numérique de drogue, un formulaire de présentation HC/SC 3011 distinct, dûment rempli, doit être soumis pour chaque formulation, concentration et forme posologique. Pour tous les autres types de présentations, seule la partie 2 doit être remplie et soumise pour chaque formulation, concentration et forme posologique.
Nota : Les renseignements complémentaires ou supplémentaires soumis à l'égard d'une présentation déjà déposée doivent être uniquement accompagnés d'une copie de la lettre de Santé Canada demandant de plus amples renseignements.
Envoi des présentations de médicaments
Médicaments à usage humain
Les demandes d'essai clinique et les modifications pour les médicaments à usage humain doivent être envoyées directement à la direction compétente, comme suit :
Produits pharmaceutiques
Bureau des essais cliniques
Direction des produits thérapeutiques
Holland Cross, tour B, 5e étage
Indice de l'adresse : 3105A
1600, rue Scott
Ottawa (Ontario)
Canada K1A 0K9
Produits biologiques/radiopharmaceutiques
Direction des produits biologiques et
des thérapies génétiques
Division des affaires réglementaires
Santé Canada
Indice de l'adresse : 0701A
200, pré Tunney
Ottawa (Ontario) K1A 0K9
Tous les autres types de présentations doivent être envoyés au Bureau des présentations et de la propriété intellectuelle, Direction de la gestion des ressources et des opérations, Santé Canada.
Médicaments à usage vétérinaire
Pour obtenir des instructions sur la manière de déposer des présentations de médicaments vétérinaires, veuillez envoyer un courriel à la Division de la gestion des présentations et du savoir à vdd.skmd.so-dgps.dmv.cp@hc-sc.gc.ca.
Partie 1 - Renseignements sur le fabricant/promoteur et le produit
Article 1-4
Directive
À l'usage de Santé Canada seulement
Article 5
Directive
Sélectionner la classe de produit. Voici les inscriptions permises :
- Humain
- Vétérinaire
- Désinfectant
Le type de présentation déposé auprès de Santé Canada. Voici les inscriptions permises :
- Demande d'essai clinique (DEC)
- Modification de demandes d'essai clinique (MDEC)
- Présentation de Drogue Expérimentale Nouvelle (PDEN)
- Modification à une Présentation de Drogue Expérimentale Nouvelle (MPDEN)
- Présentation de drogue nouvelle (PDN)
- Présentation de drogue nouvelle à usage vétérinaire (PDNV)
- Présentation de drogue nouvelle pour les produits désinfectants (PDN-D)
- Supplément à une présentation de drogue nouvelle (SPDN)
- Supplément à une présentation de drogue nouvelle à usage vétérinaire (SPDNV)
- Présentation abrégée de drogue nouvelle (PADN)
- Supplément à une présentation de drogue nouvelle - Confirmation (SPDN-c)
- Supplément à une présentation abrégée de drogue nouvelle - Confirmation (SPADN-c)
- Supplément à une présentation de drogue nouvelle pour les produits désinfectants (SPDN-D)
- Présentation abrégée de drogue nouvelle à usage vétérinaire (PADNV)
- Supplément à une présentation abrégée de drogue nouvelle (SPADN)
- Supplément à une présentation abrégée de drogue nouvelle à usage vétérinaire (SPADNV)
- Présentation de drogue nouvelle pour usage exceptionnel (EU PDN)
- Supplément à une présentation de drogue nouvelle pour usage exceptionnel (EU SPDN)
- Présentation abrégée de drogue nouvelle pour usage exceptionnel (EU PADN)
- Supplément à une présentation abrégée de drogue nouvelle pour usage exceptionnel (EU SPADN)
- Préavis de modification - pour les changements liés à la qualité des produits biologiques et des produits radiopharmaceutiques (PM)
- Préavis de modification d'un médicament à usage vétérinaire (PMV)
- Demande d'identification numérique de médicament pour un produit pharmaceutique, y compris des produits sans ordonnance attestant d'une norme d'étiquetage (DDIN) Note de bas de page 1
- Demande de DIN pour les produits biologiques (DDINB) Note de bas de page 1
- Demande de DIN pour les produits désinfectants (DDIND) Note de bas de page 1
- Demande de DIN pour un produit monographique de catégorie IV (DDINF) Note de bas de page 1
- Demande d'identification numérique de drogue à usage vétérinaire - tous les types (DINV) Note de bas de page 3
- Changement post-autorisation de Titre 1 (CPA) Note de bas de page 2
- Changement post-autorisation de Titre 1 pour les produits biologiques (CPA-B) Note de bas de page 2
- Changement post-autorisation de Titre 1, concernant des drogues à usage vétérinaire - Changement de DIN (CPAV) Note de bas de page 2
- Changement de nature administrative du nom du fabricant/produit/accords d'utilisation de licence (ADMIN)
- Changement de nature administrative du nom du fabricant/produit/accords d'utilisation de licence concernant des drogues à usage vétérinaire (VADMIN)
- COVID-19 Demande en vertu de l'arrêté d'urgence (COV19) (Ne s'applique pas aux demandes d'essais cliniques) Note de bas de page 4
- COVID-19 Modification à une demande en vertu de l'arrêté d'urgence (COV19A) (Ne s'applique pas aux demandes d'essais cliniques) Note de bas de page 4
- COVID-19 Demande en vertu de l'arrêté d'urgence (VCOV19) (Ne s'applique pas aux demandes d'essais cliniques) Note de bas de page 4
- COVID-19 Modification à une demande en vertu de l'arrêté d'urgence (COV19A) (Ne s'applique pas aux demandes d'essais cliniques) Note de bas de page 4
- Présentation de drogue nouvelle avec flexibilités - Médicament COVID-19 désigné (PDN CV)
- Présentation de drogue nouvelle à usage vétérinaire avec flexibilités - Médicament COVID-19 désigné (PDNV CV)
Article 6
Directive
Lorsqu'il y a lieu, indiquer le nombre de volumes que contient la présentation de médicament. Préciser le nombre de volumes originaux, suivi du nombre de doubles exemplaires.
Article 7
Directive
Annexe : à remplir seulement s'il s'agit d'un médicament visé à l'annexe C (produits radiopharmaceutiques) et/ou à l'annexe D (produits biologiques) de la Loi sur les aliments et drogues, à l'annexe F (médicaments d'ordonnance) du Règlements sur les aliments et drogues et veuillez préciser si le produit est visé par la Loi réglementant certaines drogues et autres substances.
Article 8
Directive
La marque nominative, marque déposée ou nom du produit est le nom distinctif attribué à ce produit par le fabricant/promoteur et sous lequel ce médicament (produit) sera vendu/annoncé. La marque nominative est aussi le nom utilisé pour identifier le produit dans toute correspondance portant sur la présentation et sur l'étiquette et la monographie du produit/notice de conditionnement, au besoin. Si le nom du produit n'est pas encore établi, p. ex. dans le cas d'une DEC, DNRV ou PDN, on peut utiliser le nom propre ou usuel du médicament ou le code de recherche.
Pour DEC, MDEC, NDRV, MNDRV, veuillez inscrire le nom du produit de recherche. Si plus d'un produit de recherche fabriqué par le promoteur est utilisé dans le cadre de l'essai clinique, il faut tous les inscrire puis remplir la partie 2 pour chacun de ces produits. Si le fabricant du produit de recherche n'est pas le promoteur de l'essai, il faut au moins remplir le champ 56. (Nota : un produit de recherche est un produit utilisé dans le cadre d'un essai clinique. Il peut s'agir d'un produit qui n'est pas disponible au Canada, d'un produit en cours d'élaboration ou encore d'un produit déjà approuvé au Canada, mais utilisé pour une indication non approuvée.)
Si après avoir soumis une présentation PDN, PADN, DIN, PDNV, PADNV ou DINV vous désirez changer le nom du produit inscrit sur le formulaire de demande, et ce, avant la fin du processus d'évaluation de cette présentation, soumettre un avis écrit du changement proposé à la même adresse où la présentation initiale a été envoyée (voir la page couverture), avec une note indiquant le numéro de la présentation et le nom original du produit. Si vous souhaitez modifier le nom du produit une fois la présentation approuvée, veuillez suivre la politique de Santé Canada intitulée Changement dans le nom d'un fabricant et/ou d'un produit.
Article 9
Directive
Le nom propre d'un produit est le nom qui lui est attribué à l'article C.01.002 du Règlement sur les aliments et drogues ou qui apparaît en caractères gras dans les autres articles du Règlement ou le nom du médicament dans sa forme finie mentionné dans le titre d'une monographie ou dans l'une des publications officielles figurant à l'annexe B de la Loi sur les aliments et drogues.
Exemples : Acétaminophène
Comprimés de sulfate ferreux
Le nom usuel est le nom sous lequel un médicament à ingrédient unique est communément connu/désigné dans les revues scientifiques ou techniques autres que les publications mentionnées à l'annexe B de la Loi sur les aliments et drogues. Le nom usuel comprend la forme pharmaceutique lorsqu'il est utilisé pour désigner le médicament dans sa forme finie.
En l'absence d'un nom propre, et si le médicament est constitué d'un seul ingrédient médicinal, inscrire le nom usuel. En l'absence d'un nom propre, et si le médicament est constitué de plus d'un ingrédient médicinal, tous les noms usuels devraient être indiqués. Dans le cadre d'un essai clinique, il faut énumérer toutes les substances actives de recherche et les séparer par une barre oblique (/).
Article Section A
Directive
Renseignements sur le fabricant/promoteur : Dans la section A (10 à 22), fournir les renseignements demandés sur le fabricant/promoteur au nom duquel la présentation de médicament est déposée et, lorsqu'une identification numérique de drogue(DIN) ou un Avis de conformité (AC) doit être délivré, la société au nom de laquelle le DIN/l'AC sera enregistrée (c.-à.-d. le détenteur du DIN/AC), et dont le nom devra apparaître sur l'étiquette et la monographie du produit/notice de conditionnement.
Pour DEC et MDEC, le titre 5 de la partie C du Règlement sur les aliments et drogues définit le promoteur comme une personne physique ou morale, un établissement ou un organisme qui mène un essai clinique. Inscrire le nom complet du promoteur au nom duquel la DEC ou la MDEC est déposée. Ne pas utiliser d'abréviations dans le nom du promoteur. Prendre note que le promoteur n'est pas nécessairement la société qui fabrique le médicament. Dans le cas de DEC et de MDEC présentées par un chercheur, si le promoteur est une personne physique, inscrire également le nom de l'établissement ou de l'organisme associé.
Dans le cas de NDRV et de MNDRV, cette définition du promoteur s'applique également.
Article 10
Directive
Code de la société : Lorsqu'il est connu, inscrire le code à quatre ou cinq chiffres attribué par Santé Canada au fabricant/promoteur, p. ex. 4567. S'il est inconnu, veuillez ne rien inscrire dans ce champ.
Article 11
Directive
Indiquer le nom légal complet du fabricant/promoteur au nom duquel la présentation de médicament à traiter est déposée et, le cas échéant, au nom duquel le DIN/l'AC doit être enregistré. Ne pas utiliser d'abréviations dans le nom de la société. Prendre note que le fabricant/promoteur n'est pas nécessairement la société qui fabrique le médicament. Veuillez éviter l'usage de formules telles que « faisant affaire comme » dans le nom du fabricant/promoteur, par exemple Hamilton Frères faisant affaire comme Famille Hamilton. Il devrait opter pour « Hamilton Frères » ou « Famille Hamilton », ou encore « Hamilton Frères, une Division de Famille Hamilton ».
Si après avoir soumis une présentation PDN, PADN, DIN, PDNV, PADNV ou DINV, vous désirez changer le nom du fabricant/promoteur, et ce, avant la fin du processus d'évaluation de la présentation, soumettre un avis écrit du changement proposé à la même adresse où la présentation initiale a été envoyée (voir la page couverture) avec une note indiquant le numéro de la présentation et le nom original du fabricant/promoteur. Si vous désirez modifier le nom du fabricant/promoteur une fois la présentation approuvée, veuillez suivre la politique de Santé Canada intitulée Changement dans le nom d'un fabricant et/ou d'un produit. Il faut également noter qu'une présentation de changement de nature administrative qui renvoie à une PDN, PADN, PDNV ou PADNV originale doit être soumise et approuvée avant tout changement de nom du fabricant/promoteur pour une PSDN, une PSADN, un PM, une PSDNV, une PSADNV ou un PMV déjà soumis.
Pour DEC et MDEC, le titre 5 de la partie C du Règlement sur les aliments et drogues définit le promoteur comme une personne physique ou morale, un établissement ou un organisme qui mène un essai clinique. Indiquer le nom légal complet du promoteur au nom duquel la DEC ou la MDEC est déposée. Ne pas utiliser d'abréviations dans le nom du promoteur. Prendre note que le promoteur n'est pas nécessairement la société qui fabrique le médicament. Dans le cas de DEC et de MDEC déposées par un chercheur, si le promoteur est une personne physique, inscrire également le nom de l'établissement ou de l'organisme associé.
Dans le cas de NDRV et de MNDRV, cette définition du promoteur s'applique également.
Article 12-16
Directive
Indiquer l'adresse postale complète du fabricant/promoteur identifié au champ 11. Si une adresse civique est utilisée, indiquer le numéro de bureau/suite (le cas échéant), ainsi que la rue et le numéro (12), la ville (13), la province/État (14), le pays (15) et le code postal ou le code de zone (16). L'usage d'une boîte postale au lieu du nom de la rue/suite pour décrire l'adresse du fabricant/promoteur n'est pas permis.
Article 17-22
Directive
Indiquer le nom du principal contact (17) travaillant à l'adresse (12 à 16) du fabricant/promoteur identifié au champ 11 et fournir les renseignements nécessaires pour communiquer avec cette personne, c.-à-d. les numéros de téléphone et de télécopieur (18 à 19), le poste/titre (21), l'adresse électronique (22) et, le cas échéant, la langue préférée (20).
Les besoins opérationnels et ceux du système exigent que ce nom soit le même pour tous les DIN enregistrés au nom du fabricant/promoteur identifié au champ 11 lorsque la société détient plus d'un DIN. Il faut noter qu'il ne s'agit PAS nécessairement du contact pour la présentation de médicament à traiter, mais du principal contact pour le fabricant/promoteur à l'adresse indiquée.
En raison de l'objectif de rendement à court terme associé aux essais cliniques au Canada, cette section comprend un autre contact dans le cas où on ne peut pas joindre le contact mentionné à la section B.
Article Section B
Directive
Contact pour la présentation de médicament à traiter : Les renseignements fournis à la section B (23 à 34) concernent la personne à contacter spécifiquement pour la présentation de médicament à traiter, c.-à-d. la personne/société à qui Santé Canada doit adresser toute correspondance ayant trait à la présentation en question. Pour éviter toute confusion, il faut remplir la section B même si le contact pour la dite présentation est mentionné dans un autre champ pour un autre rôle.
Article 23
Directive
Inscrire le nom de la société à laquelle appartient le contact pour la présentation de médicament (c.-à-d. où il est employé). Ne pas utiliser d'abréviations dans le nom de la société. Si le contact n'appartient à aucune société, inscrire son nom.
Article 24-28
Directive
Inscrire l'adresse de la société identifiée au champ 23. Si une adresse civique est utilisée, indiquer le numéro de bureau/suite (le cas échéant), ainsi que la rue et le numéro (24), la ville (25), la province/État (26), le pays (27) et le code postal ou le code de zone(28). Mentionner le numéro de la boîte postale (24) le cas échéant.
Article 29-34
Directive
Indiquer le nom de la personne à contacter au sujet de la présentation de médicament à traiter (29), c.-à-d., le nom de la personne à qui Santé Canada doit adresser toute correspondance au sujet de la présentation de médicament à traiter. Fournir les renseignements nécessaires pour communiquer avec cette personne, soit les numéros de téléphone et de télécopieur (30 et 31), le poste/titre (33), l'adresse électronique (34) et, le cas échéant, la langue préférée (32). Veuillez noter que le choix du mode de correspondance revient à Santé Canada.
Article Section C
Directive
Adresse pour le courrier réglementaire : Dans la section C (35 à 46), fournir les renseignements nécessaires pour indiquer où et à qui Santé Canada doit envoyer le courrier réglementaire autre que le courrier concernant spécifiquement la présentation de médicament à traiter (section B), p. ex. le formulaire de déclaration annuelle, les avis de modification à des règlements ou politiques qui s'appliquent aux DIN enregistrés au nom du fabricant/promoteur identifié au champ 11. Cocher « même que A ci-dessus » ou, s'il s'agit d'une adresse différente, remplir les champs 35 à 46. Les besoins opérationnels et ceux du système exigent que le nom/l'adresse pour le courrier réglementaire soient les mêmes pour tous les DIN enregistrés au nom du fabricant/promoteur identifié au champ 11 lorsque la société détient plus d'un DIN.
Pour DEC, MDEC, NDRV et MNDRV, cocher S.O. si la section C si cela ne s'applique pas. Si la case S.O. est cochée, les champs restants devraient être vides.
Article 35
Directive
Inscrire le nom complet de la société qui agira au nom du fabricant/promoteur identifié au champ 11 pour ce qui concerne le courrier réglementaire envoyé par Santé Canada, p. ex. déclaration annuelle, avis de modification à des règlements ou politiques qui s'appliquent aux DIN enregistrés au nom du fabricant/promoteur indiqué au champ 11.
Article 36-40
Directive
Inscrire l'adresse de la société mentionnée au champ 36. Si une adresse civique est utilisée, indiquer le numéro de bureau/suite (le cas échéant), ainsi que la rue et le numéro (36), la ville (37), la province/État (38), le pays (39) et le code postal ou le code de zone (40). Mentionner le numéro de la boîte postale (36) le cas échéant.
Article 41-46
Directive
Préciser le nom du principal contact travaillant à l'adresse mentionnée aux champs 36 à 40 et les renseignements nécessaires pour communiquer avec cette personne, c.-à-d. ses numéros de téléphone et de télécopieur (42 et 43), son poste/titre (45), son adresse électronique (46) et, le cas échéant, la langue préférée (44). Veuillez noter que le choix du mode de correspondance revient à Santé Canada.
Article Section D
Directive
Importateur canadien est responsable de la vente de ce produit au Canada. Remplir la section D (47 à 52) seulement si l'adresse du fabricant/promoteur mentionnée aux champs 11 à 16 n'est pas située au Canada. Si la société est la même que celle mentionnée au champ 35, cocher « même que C ci-dessus » ou, s'il s'agit d'une société différente, remplir chaque partie de la section D.
Sauf pour DEC et MDEC, si les drogues pour les essais cliniques sont importées au Canada, les importateurs devraient être autorisés par le promoteur, quel que soit l'emplacement du promoteur. L'annexe 1 devrait être remplie et soumise pour chaque importateur au Canada. Énumérer chaque importateur concerné autorisé à importer la drogue nouvelle aux fins de l'essai clinique indiqué dans la demande. Les importateurs canadiens doivent être situés au Canada. Pour plus de renseignements, consulter la Ligne directrice à l'intention des promoteurs d'essais cliniques. À mesure que d'autres importateurs sont identifiés, des copies supplémentaires de l'annexe 1 devront être fournies à Santé Canada. Si l'importateur est resté le même au moment où une modification à une demande d'essai clinique (MDEC) est soumise, il n'est pas nécessaire de soumettre de nouveau l'annexe 1.
Article 47
Directive
Inscrire le nom complet de l'importateur canadien. Les importateurs canadiens doivent être situés au Canada.
Article 48-52
Directive
Inscrire l'adresse de la société mentionnée au champ 47. Si une adresse civique est utilisée, indiquer le numéro de bureau/suite (le cas échéant), ainsi que la rue et le numéro (48), la ville (49), la province/État (50), et le code postal (52). Veuillez ne pas modifier le nom du pays « Canada » au champ 51.
Article Section E
Directive
Si un DIN ou un AC doit être délivré, indiquer l'adresse parmi celles susmentionnées où le formulaire de déclaration de médicament ou l'AC doit être envoyé, c.-à-d. l'adresse de la section A, B, C ou D. Cocher toutes les options applicables.
Article 53
Directive
Lorsque la présentation de médicament soumise comporte des renvois à des présentations de médicaments connexes (p. ex. à une DEC précédant une MDEC, une DEC précédant une PDN, une PDN concernant une DEC, la PDN ascendante pour une PSDN ou une DIN précédant une MPAT), indiquer le type de présentation (voir au champ 5 ci-dessus), le numéro de contrôle (de la présentation), la marque nominative du médicament, le nom du fabricant/promoteur de la présentation connexe, le numéro de dossier de la présentation connexe, la date d'approbation, les DIN connexes (le cas échéant) et la raison de la présentation connexe (p. ex. brève description du protocole d'une DEC, raisons expliquant la présentation supplémentaire, etc.).
Il faudrait indiquer le nom propre, usuel ou générique si la marque nominative n'est pas connue au moment de soumettre la demande. Seules les demandes mentionnées dans la présentation de médicament à traiter devraient être indiquées sous cette section. Reproduire et remplir le champ 53 au complet, au besoin.
DIN connexe(s) : Indiquer les DIN des produits (le cas échéant) qui sont directement visés par la présente demande. Cela permettra de repérer le produit afin d'y apporter toute modification, p. ex., mise à jour de la monographie de produit ou de tout autre renseignement dans la Base de données sur les produits pharmaceutiques (BDPP). Si aucun DIN n'a été émis pour le produit, veuillez inscrire S.O. pour sans objet.
Partie 2 - Renseignements sur la formulation du produit pharmaceutique
Article 54
Directive
La date de péremption/durée de conservation recommandée correspond à la période, en années et en mois, pendant laquelle le produit pharmaceutique conserve son activité, sa pureté et ses caractéristiques physiques indiquées sur l'étiquette.
Pour DEC, MDEC, NDRV et MNDRV, il n'est pas nécessaire de remplir ce champ si le produit à utiliser pour l'essai clinique est commercialisé au Canada ou si le produit de recherche est utilisé dans le cadre d'une étude de bioéquivalence/biodisponibilité.
Article 55
Directive
Inscrire le nom du ou des pays où la forme posologique finale du médicament est fabriquée. Par exemple, si la forme posologie du produit pharmaceutique est un comprimé ou une solution, indiquer le ou les pays où le comprimé ou la solution a été fabriqué(e). Cela n'est pas nécessairement le ou les pays où le produit a été emballé/étiqueté.
Pour DEC, MDEC, NDRV et MNDRV, veuillez indiquer S.O. si le fabricant du produit de recherche n'est pas le promoteur de l'essai.
Article 56
Directive
Indiquer le ou les ingrédients médicinaux (actifs) qui contribuent à l'utilisation proposée du produit par son nom ou leurs noms propres ou usuels. Lorsque la norme de fabrication d'un ingrédient est conforme à une norme officinale mentionnée à l'annexe B, utiliser l'abréviation de cette pharmacopée, p. ex. United States Pharmacopeia (USP), British Pharmacopoeia (BP). Lorsque les spécifications régissant la fabrication de l'ingrédient s'écartent et excèdent ou sont équivalentes à la norme officinale, la norme du fabricant (Mfr Std) peut être indiquée. Veuillez inscrire S.O. si aucune norme officinale n'existe pour l'ingrédient actif.
La concentration de l'ingrédient médicinal (actif) doit être exprimée comme suit :
- Formes pharmaceutiques discrètes (p. ex. comprimés) - g ou mg (title tag = milligramme) / forme pharmaceutique
- Poudre pour administration orale - g ou mg/ml, g ou mg / unité posologique (p. ex./5 ml)
- Liquide pour administration parentérale - mg/ml ou %
- Liquide pour administration orale - g ou mg/ml, g ou mg / unité posologique (p. ex./5 ml)
- Crème, pommade, lotion, etc. - mg or mL/g, g or mg/mL or %
- Poudre pour solution, etc. - unité/unité posologique (p. ex. /flacon de 20 ml)
| Numéro CAS (le cas échéant) | Nom de l'ingrédient | Norme | Norme Concentration | Unité | Par | Calculé sous forme de base? | Source humaine ou animale | ||
|---|---|---|---|---|---|---|---|---|---|
| 564-25-0 | Doxycycline (sous-forme d'hyclate de doxycycline) | USP | 100 | mg | comprimé | 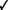 Oui Oui |
Non | Oui | Non |
| 86541-74-4 | Chlorhydrate de bénazépril | USP | 5 | mg | comprimé | Oui | Non |
Oui | Non |
| s.o. | Antithrombine III (humain) | s.o. | 1350 - 1650 | IU | flacon | Oui | Non |
Oui |
Non |
Dans les exemples qui précèdent, la concentration de la molécule active de doxycycline (présenté sous forme d'hyclate de doxycycline) est déclarée et donc calculée sous forme de base. Dans le cas du chlorhydrate de bénazépril, c'est la concentration du sel qui est déclarée, et donc cette concentration n'est pas calculée sous forme de base.
Indiquer si l'ingrédient provient d'une source animale ou humaine. Si l'ingrédient provient d'une source animale ou humaine, veuillez remplir et soumettre l'annexe 4.
En ce qui concerne les demandes d'identification numérique de drogues, il faut remplir et soumettre un formulaire HC/SC 3011 distinct pour chaque formulation et concentration. Pour tous les autres types de présentations, seule la partie 2 doit être reproduite, remplie et soumise pour chaque formulation et concentration. Si tous les renseignements sont les mêmes pour les champs restants, il n'est donc pas nécessaire de reproduire ces champs pour chaque concentration/forme posologique.
Par exemple, si la demande vise un produit régi par le titre 8 (nouvelle drogue) offert en deux concentrations (c.-à-d. 5 mg et 10 mg) et que la concentration est la seule différence touchant la partie 2, les champs 56 et 57 devraient donc être reproduits.
Article 57
Directive
Énumérer les ingrédients non médicinaux de la même façon que les ingrédients médicinaux (actifs) au champ 56 ci-dessus. Il n'est cependant pas nécessaire d'indiquer si l'ingrédient est calculé sous forme de base ou de sel. Inscrire d'abord les agents de conservation ensuite les colorants. Le reste des ingrédients non médicinaux devraient être indiqués au champ C - Autre et, le cas échéant, indiquer les ingrédients utilisés dans l'enveloppe des capsules au champ D. Dans le cas des produits régis uniquement par le titre 1, décrire le but des ingrédients non médicinaux inscrits dans la catégorie « Autre »sur une feuille distincte.
Pour les variations de formulation relatives à un même DIN (p. ex. saveurs, couleurs et parfums différents), veuillez indiquer le nom de la variante et inscrire tous les ingrédients non médicinaux pour chaque type de variante.
Indiquer si l'ingrédient provient d'une source animale ou humaine. Si l'ingrédient provient d'une source animale ou humaine, veuillez remplir et soumettre l'annexe 4.
Pour DEC, MDEC, NDRV et MNDRV, il n'est pas nécessaire de remplir ce champ si le produit à utiliser pour l'essai clinique est commercialisé au Canada ou si le produit de recherche est utilisé dans le cadre d'une étude de bioéquivalence/biodisponibilité.
Article 58
Directive
Énumérer le matériel d'origine animale/humaine pour chaque ingrédient médicinal et/ou matériel utilisé à une étape ou une autre de la fabrication du produit y compris l'excipient ou l'additif provenant de tissus d'origine humaine et/ou animale.
Indiquer le numéro CAS (le cas échéant), le nom du matériel et la norme pour le matériel d'origine humaine/animale utilisé dans la fabrication du produit. Sélectionner l'option Oui dans la colonne « Présence dans le récipient final » si l'ingrédient est présent dans le récipient final (selon la norme de qualité applicable au produit).
En ce qui concerne les demandes d'essai clinique pour les produits pharmaceutiques, il ne faut pas soumettre l'annexe 4 étant donné que les attestations ou les renseignements requis sont indiqués dans la demande.
Article 59
Directive
Faire le choix approprié selon la définition pratique de nanomatériaux de Santé Canada :
Santé Canada estime que toute substance ou tout produit fabriqué et toute constitutive ou structure ou tout ingrédient ou dispositif constitue un nanomatériau :
- s'il est à l'échelle nanométrique, ou dans les limites de celle-ci, dans au moins une dimension externe ou présente une structure interne ou en surface l'échelle nanométrique; ou,
- s'il est plus petit ou plus grand que l'échelle nanométrique dans toutes les dimensions et affiche un ou plusieurs phénomène ou propriétés à l'échelle nanométrique.
Aux fins de la présente définition :
- le terme « à l'échelle nanométrique » signifie 1 à 100 nanomètres inclusivement;
- le terme « propriétés ou phénomène à l'échelle nanométrique » signifie des propriétés qui sont attribuables à la taille à aux effets; ces propriétés sont faciles à distinguer des propriétés chimiques ou physiques des atomes, molécules et matériaux particuliers et les matériaux en vrac; et,
- le terme « fabriqué » comprend les processus techniques et les contrôles de la matière.
Article 60
Directive
Indiquer la forme posologique (forme pharmaceutique) proposée du produit, p.ex. comprimé, capsule, crème, poudre pour solution, etc.
En ce qui concerne les demandes d'identification numérique de drogues, il faut remplir et soumettre un formulaire HC/SC 3011 distinct pour chaque forme posologique. Pour tous les autres types de présentations, seule la partie 2 doit être reproduite, remplie et soumise pour chaque forme posologique.
Article 61
Directive
Indiquer le type de récipient(s) qui sera utilisé et le ou les formats proposés, p. ex. flacon de 100 comprimés, tube de 50 g, fiole de 5 ml.
Pour DEC, MDEC, NDRV et MNDRV, veuillez indiquer S.O. étant donné qu'il n'est pas nécessaire de remplir ce champ si les renseignements sont inconnus au moment de soumettre la demande.
Article 62
Directive
Inscrire la classification thérapeutique et pharmacologique appropriée du médicament, p. ex. bloqueurs des canaux calciques, agent de modification de la réponse biologique, antagonistes du récepteur H2 de l'histamine, désinfectant.
Article 63
Directive
Indiquer la ou les voies d'administration proposées, p. ex. voie orale, voie intraveineuse, application topique, etc. Pour une demande de DIN concernant un désinfectant, l'information recueillie dans cette section porte sur son usage tel que mentionné au champ 65; le promoteur peut donc inscrire « Voir champ 65 » au lieu d'indiquer l'usage du désinfectant.
Article 64
Directive
Indiquer si le médicament est un produit biologique, un produit radiopharmaceutique, un produit pharmaceutique, un désinfectant ou la combinaison d'un médicament et d'un instrument médical. S'il s'agit d'une combinaison d'un médicament et d'un instrument médical, préciser également de quel type de médicament il est question, p. ex. biologique/radiopharmaceutique ou pharmaceutique.
Article 65
Directive
Indiquer s'il s'agit d'un médicament à usage humain (population pédiatrique ou adulte - sélectionner les deux choix le cas échéant) ou à usage vétérinaire ou qui doit servir comme désinfectant ou produit radiopharmaceutique.
Population pédiatrique est un terme défini comme suit dans le Règlement sur les aliments et drogues (C.08.004.1) : les bébés prématurés nés avant la 37e semaine de gestation, les bébés nés à terme et âgés de 0 à 27 jours, tous les enfants âgés de 28 jours à deux ans, ceux âgés de deux ans et un jour à 11 ans, et enfin ceux âgés de 11 ans et un jour à 18 ans.
Si le produit est un désinfectant, indiquer s'il est destiné à être utilisé en milieu hospitalier; sur des surfaces servant à la préparation d'aliments; pour la désinfection/stérilisation d'instruments médicaux; en milieu agricole; en milieu domestique; dans un établissement/ une industrie; ou pour des lentilles cornéennes.
Article 66
Directive
S'il s'agit d'un médicament en vente libre (à usage humain et/ou vétérinaire) qui correspond à une ou plusieurs allégations énumérées à l'annexe A, veuillez remplir et soumettre l'annexe 5. Pour obtenir de plus amples renseignements et instructions, veuillez consulter le document d'orientation intitulé : Ébauche de la ligne directrice - Annexe A et article 3 de la Loi sur les aliments et drogues.
Article 67
Directive
Préciser l'indication/utilisation proposée du médicament, p. ex. pour le traitement de l'angine de poitrine et de l'hypertension, pour traiter les symptômes du rhume des foins, pour la prophylaxie de l'infection par le virus de la rougeole.
Pour un nouveau médicament, l'indication/utilisation proposée devrait être un résumé de l'indication décrite dans la monographie de produit. Il faudrait utiliser une terminologie scientifique clé pour cette section.
Pour un produit désinfectant, inclure une ou les deux options suivantes : 1) Désinfectant pour surfaces dures; 2) Désinfectant pour lentilles cornéennes. Les propositions d'indications telles que « désinfectant /assainisseur » ou « nettoyant pour cuvette » ne sont pas permises.
Article 68
Directive
Préciser la posologie proposée du produit médicamenteux, y compris la dose quotidienne maximale, p. ex. un comprimé quatre fois par jour.
Article 69
Directive
Confirmer que les ébauches des étiquettes (intérieures et extérieures) destinées à être utilisées au Canada (y compris la notice de conditionnement, le cas échéant) sont incluses dans la présentation L'ébauche des étiquettes ne comprend pas la monographie du produit.
Pour DEC, MDEC, NDRV et MNDRV, les étiquettes n'ont pas à être soumises, sauf sur demande de la direction compétente.
Article 70
Directive
Ne remplir ce champ que si la présentation de médicament à traiter est une PSDN, une PSADN (tous les types de médicaments à usage humain), une PSDNV, une PSADNV (médicament à usage vétérinaire) ou une demande de DIN pour un produit biologique. Indiquer la raison du dépôt de la présentation (p. ex. nouvelle voie d'administration ou nouvelle forme pharmaceutique, nouvelle concentration, etc.). Cocher toutes les options applicables.
Article 71
Directive
Ne remplir ce champ que si la présentation de médicament en question est un préavis de modification concernant une drogue nouvelle (à usage humain ou vétérinaire). Conformément à la politique de Santé Canada intitulée Lignes directrices sur les changements survenus après l'avis de conformité (AC), indiquer toutes les modifications visées par la présentation.
Article 72-74
Directive
Remplir les champs 72 à 74 uniquement dans le cas de médicament à usage vétérinaire.
Article 72
Directive
Préciser les espèces et les sous-types auxquels le produit médicamenteux est destiné, p. ex. volaille (espèce) - poules pondeuses (sous-type).
Article 73
Directive
Indiquer si le médicament est prévu pour utilisation chez des animaux producteurs d'aliments.
Article 74
Directive
Indiquer le délai d'attente proposé pour chaque espèce mentionnée au champ 72.
Article 75
Directive
Indiquer, en caractères d'imprimerie, le nom de la personne autorisée à signer le formulaire par le fabricant/promoteur mentionné au champ 11; la personne qui signe le formulaire atteste que les renseignements fournis dans le formulaire sont conformes aux désirs du fabricant/promoteur.
Article 76-77
Directive
La signature de la personne autorisée (76) et la date de signature du formulaire (année/mois/jour) (77).
Article 78-80
Directive
Le titre du poste occupé par le signataire autorisé (78), ainsi que les numéros de téléphone et de télécopieur de ce dernier (79 à 80).
Article 81
Directive
Le nom de la société à laquelle le signataire autorisé appartient. Si le signataire est une tierce personne qui appartient à une société autre que celle du fabricant/promoteur identifié au champ 11, une lettre d'autorisation signée par le fabricant/promoteur (champ 11) doit être soumise avec le formulaire de présentation de médicament (voir annexe 2).
Article Annexe 1
Directive
Remplir l'annexe 1 (ou une autorisation semblable) uniquement pour les demandes d'essai clinique et les modifications à une demande d'essai clinique. Il faut remplir et soumettre le formulaire si le promoteur de l'essai clinique à autorisé un ou plusieurs tiers d'importer des drogues pour l'essai clinique au Canada. Il faut identifier chaque importateur autorisé à importer la drogue nouvelle au Canada aux fins de l'essai clinique décrit dans cette demande. À mesure que d'autres importateurs sont identifiés, des copies supplémentaires de l'annexe 1 devront être fournies à Santé Canada. Si l'importateur est le même au moment où une demande de modification est soumise, il est inutile de soumettre de nouveau l'annexe 1.
Chaque protocole doit être accompagné d'un formulaire HC/SC 3011 distinct et de l'annexe 1 correspondante.
Article Annexe 2
Directive
Remplir l'annexe 2 (ou une autorisation semblable) uniquement si le signataire du formulaire HC/SC 3011 est une tierce partie agissant pour le compte du fabricant/promoteur de la société identifiée au champ 11. Noter qu'une autorisation distincte est requise pour chaque demande.
Article Annexe 3
Directive
Remplir l'Annexe 3 - Renseignements sur la demande d'essai clinique uniquement pour les demandes d'essai clinique et les modifications à une demande d'essai clinique. Le formulaire HC/SC 3011 doit être accompagné de l'annexe 3, signée et datée, au moment de présenter un essai clinique ou une modification (clinique ou relative à la qualité) d'un essai clinique déjà approuvé.
Article 82
Directive
Indiquer le numéro de protocole de l'essai clinique. Veuillez noter que le numéro de protocole devrait être le même pour la durée de l'essai. On peut indiquer le numéro de version et de modification du protocole soumis au moment de la présentation de la demande. Cependant, le numéro de protocole original devrait toujours être indiqué.
Article 83
Directive
Indiquer le titre du protocole d'essai clinique.
Article 84
Directive
Dans le cas d'un produit de recherche concerné dans l'essai clinique, indiquer si le produit commercialisé au Canada est utilisé. Si le produit commercialisé au Canada est utilisé, veuillez indiquer l'identification numérique de drogue (DIN). Si un produit commercialisé à l'étranger est utilisé, indiquer le ou les pays desquels le produit a été obtenu. Si le produit de recherche n'est pas commercialisé, inscrire S.O.
Article 85
Directive
Indiquer si l'on prévoit la participation à l'essai clinique de la population pédiatrique, de 0 à 18 ans (sexe féminin ou masculin) et de population adulte (femmes ou hommes).
Population pédiatrique est un terme défini comme suit dans le Règlement sur les aliments et drogues (C.08.004.1) : les bébés prématurés nés avant la 37e semaine de gestation, les bébés nés à terme et âgés de 0 à 27 jours, tous les enfants âgés de 28 jours à deux ans, ceux âgés de deux ans et un jour à 11 ans, et enfin ceux âgés de 11 ans et un jour à 18 ans.
Cocher toutes les options applicables.
Article 86
Directive
Indiquer sur quelle phase de l'essai clinique porte la demande. Pour de plus amples renseignements, veuillez consulter la Ligne directrice à l'intention des promoteurs concernant les demandes d'essais cliniques.
Article 87
Directive
Si le comité d'éthique pour la recherche a refusé d'approuver le protocole d'essai clinique et/ou le formulaire de consentement éclairé, confirmer que l'information mentionnée dans la Ligne directrice à l'intention des promoteurs d'essais cliniques est envoyée avec la demande. Si aucun comité d'éthique pour la recherche n'a refusé d'approuver le protocole ou le formulaire de consentement éclairé, cocher la case S.O. pour sans objet. Si la réponse du comité d'éthique pour la recherche n'est pas encore connue, cocher la case « Pas encore connu ».
Article 88
Directive
Un formulaire de renseignements sur le lieu de l'essai clinique devrait être joint pour chaque site d'essai clinique connu proposé au moment de soumettre la demande d'essai clinique ou de modification. Veuillez ne pas présenter de formulaire avant d'avoir rempli tous les champs, y compris le champ 37 et/ou 45 du formulaire de renseignements sur le lieu de l'essai clinique.
Lorsque l'information sur un site d'essai clinique est obtenue une fois la demande traitée, un formulaire de renseignements sur le lieu de l'essai clinique dûment rempli doit être envoyé à la direction compétente avant le commencement de l'essai.
Article 89-90
Directive
Inscrire en caractères d'imprimerie le nom de l'agent médical ou scientifique principal au Canada (89), son numéro de téléphone et son adresse (90). Il s'agit d'un agent médical ou scientifique résidant au Canada, qui représente le promoteur et qui est responsable de fournir une attestation en regard à une DEC ou à une MDEC au moment où le formulaire est soumis, tel qu'indiqué à l'annexe 3.
Article 91-92
Directive
L'agent médical ou scientifique au Canada (91) doit signer la demande et indiquer la date de la signature (année/mois/jour) (92).
Article 93-94
Directive
Inscrire en caractères d'imprimerie le nom du cadre administratif supérieur (93) du promoteur et son numéro de téléphone (94).
Pour les essais cliniques entrepris par les inspecteurs/établissements, le chef de service compétent peut signer au nom du cadre administratif supérieur.
Article 95-96
Directive
Le cadre administratif supérieur du promoteur doit signer la demande (95) et indiquer la date de la signature (année/mois/jour) (96).
Article Annexe 4
Directive
Si vous avez répondu « Non » à toutes les questions des champs 56, 57 et 58 pour indiquer qu'aucun ingrédient de source humaine et/ou animale n'a été utilisé, veuillez ne pas remplir cette annexe.
Veuillez remplir une annexe 4 distincte pour chaque ingrédient et/ou matériel utilisé à une étape ou à une autre de la fabrication du médicament identifié comme étant de source animale et/ou humaine à la « Partie 2 - Renseignements sur la formulation du produit pharmaceutique » du formulaire de présentation de médicament.
Pour les produits biologiques, on peut soumettre des renseignements sur la source humaine/animale d'un antigène précis sous un produit de marque comme souche puis faire un renvoi avec ceux dans les demandes subséquentes pour différents produits de marque qui contiennent les mêmes antigènes. Dans ce cas, le promoteur doit indiquer les renseignements des références croisées en indiquant le nom de la marque, les DIN et le nom de l'antigène sur une page distincte au lieu de remplir le formulaire sur les tissus d'origine humaine/animale.
Il n'est pas nécessaire de soumettre de nouveau une annexe 4 pour toute présentation subséquente pour le même produit s'il n'y a aucune modification dans les renseignements médicinaux et non médicinaux et leur source.
La demande de renseignements supplémentaires sur les ingrédients d'origine animale vise à recueillir les données pertinentes sur la formulation des produits et d'utiliser ces renseignements à tout moment au cours du cycle de vie du produit, au besoin.
Article 97
Directive
Inscrire le matériel et ingrédients médicinaux et non médicinaux de source animale et/ou humaine utilisés à une étape ou à une autre de la fabrication du médicament par leur nom propre ou usuel.
Remplir une annexe 4 distincte pour chaque matériel et/ou ingrédient médicinal utilisé à une étape ou une autre de la fabrication du produit identifié comme étant de source animale et/ou humaine.
Article 98
Directive
Indiquer si le matériel/ingrédient de source animale et/ou humaine est utilisé comme ingrédient médicinal (actif), ingrédient non médicinal ou matériel utilisé à une étape ou à une autre de la fabrication du médicament.
Article 99
Directive
Préciser si l'ingrédient est de source humaine et/ou animale. Pour les animaux, veuillez cocher la case correspondante et préciser les espèces dans la catégorie indiquée. Par exemple, si l'animal est un cervidé, préciser si c'est un orignal, un cerf, un wapiti, etc.
Population contrôlée : Préciser si l'ingrédient provient d'un animal ou d'animaux appartenant à une population exclusive spécifiquement élevée dans le but de contrôler les maladies ou à des fins d'utilisations par l'industrie pharmaceutique (p. ex., certification pertinente ou normes de reconnaissance par des tiers ou des tierces parties nationales ou internationales).
Animal issu de la biotechnologie : Préciser si l'ingrédient provient d'un animal ou d'animaux issus de la biotechnologie.
Le Règlement sur la santé des animaux que régit la Loi sur la santé des animaux définit la biotechnologie comme l'« application des sciences ou de l'ingénierie à l'utilisation directe ou indirecte des organismes vivants ou de leurs parties ou produits, sous leur forme naturelle ou modifiée ». L'expression « animaux issus des biotechnologies » est une extension de la définition de biotechnologie. Elle désigne des animaux qui sont le résultat de l'application de méthodes biotechnologiques. Pour de plus amples renseignements, veuillez consulter le Cadre d'évaluation des risques zoosanitaires pour les animaux issus de la biotechnologie de l'Agence canadienne d'inspection des aliments.
Article 100
Directive
Préciser les tissus et les fluides qui sont utilisés pour obtenir les ingrédients.
Article 101
Directive
Indiquer l'âge de l'animal (en mois) au moment du prélèvement/obtention du tissu/fluide/macromolécule. Il est acceptable de fournir l'intervalle d'âge en mois.
Article 102
Directive
Indiquer tous les pays où l'animal est né et a vécu.
L'Organisation mondiale de la santé animale (OIE) attribue aux pays une classification du statut sanitaire concernant l'évaluation des risques de l'encéphalopathie spongiforme bovine (ESB). Si le pays d'origine du matériel animal utilisé dans un produit médicamenteux est classifié comme un « risque maîtrisé à l'égard de l'ESB » ou de « risque indéterminé d'ESB », le produit sera sujet à un examen additionnel sur la chimie et fabrication par Santé Canada. Pour plus de renseignements concernant l'évaluation des risques de l'encéphalopathie spongiforme bovine (ESB), veuillez consulter le site internet de l'OIE à www.oiet.int.
Article Annexe 5
Directive
Veuillez noter qu'il faut remplir le présent formulaire UNIQUEMENT dans les cas de produits vendus sans ordonnance à l'exception des produits de santé naturels pour lesquels il existe des allégations concernant des troubles médicaux inscrits à l'annexe A. Autrement dit, si la case OUI a été cochée du champ 66 du formulaire, veuillez remplir et soumettre l'annexe 5. Pour de plus amples renseignements et directives sur l'annexe A, veuillez consulter l'Ébauche de la ligne directrice - Annexe A et article 3 de la Loi sur les aliments et drogues.
Article 103
Directive
Indiquer le fabricant/promoteur tel qu'indiqué au champ 11 ci-dessus.
Article 104
Directive
Indiquer le nom du produit tel qu'indiqué au champ 8 ci-dessus.
Article 105
Directive
Déterminer l'identification numérique de drogue (DIN) si celui-ci a déjà été émis.
Article 106
Directive
Vérifier tous les troubles/maladies visés par les allégations.
Article 107
Directive
Énumérer les allégations/indications inscrites à l'annexe A associées au produit indiqué au champ 104.