
Nitrosamine impurities in medications: Guidance

Download in PDF format
(1.37 MB, 52 pages)
Organization: Health Canada
Date published: 2022-04-04
Date updated: 2025-08-01
Evaluating and managing the risks of N-nitrosamine impurities in human pharmaceutical, biological and radiopharmaceutical products
On this page
- Background
- General
- Safety
- Quality
- Appendices
- Appendix 1: Established Acceptable Intake (AI) limits for N-nitrosamine impurities
- Appendix 2: Guidance with respect to nitrosamine impurities and risk assessments for Post NOC Changes of new drug products containing chemically synthesized and semi-synthetic APIs (updated)
- Appendix 3: Enhanced Ames assay test conditions
- Appendix 4: Carcinogenic Potency Categorization Approach (CPCA) for N-nitrosamines
- Appendix 5: Bioequivalence studies for reformulated drug products (new)
Background
This guidance represents Health Canada's current thinking and recommendations on issues related to N-nitrosamine impurities (nitrosamine impurities or nitrosamines). This guidance may be subject to change as new information becomes available and if further guidance is needed for applicants and market authorization holders (MAHs).
A questions-and-answers (Q&A) document on nitrosamines was issued to MAHs on November 26, 2019. This document has undergone a number of revisions and has been further updated as a guidance document and to provide additional details to active pharmaceutical ingredient (API) manufacturers, drug product manufacturers, MAHs and importers of APIs and drug products.
In this guidance document, changes from the previous version are identified with the descriptors "new" or "updated" (as applicable). Information on a similar theme is grouped together under general headings (for example, General, Safety and Quality).
Queries about the Health Canada letters noted below can be directed as follows:
- "Information to MAHs of Human Pharmaceutical Products Regarding Nitrosamine Impurities: Request to evaluate the risk of the presence of nitrosamine impurities in human pharmaceutical products containing chemically synthesized active pharmaceutical ingredients" (October 2, 2019)
- Email to bpsenquiries@hc-sc.gc.ca
- "Information to MAHs of Human Pharmaceutical Products Regarding Nitrosamine Impurities: Request to evaluate the risk of the presence of nitrosamine impurities in biologics and radiopharmaceuticals" (December 15, 2020)
- Email to brdd.nitrosamines.dmbr@hc-sc.gc.ca
If you have queries about this guidance document, you may send an email to bpsenquiries@hc-sc.gc.ca.
General
Scope and responsibilities
1. Drug products that are within the scope of Health Canada's call for review
The request in Health Canada's call for review to evaluate the risk of the presence of nitrosamine impurities outlined in the October 2, 2019, letter applies to human pharmaceutical products with a drug identification number (DIN) containing chemically synthesized and semi-synthetic APIs. This includes:
- prescription and non-prescription (over-the-counter) drug products
- chemically synthesized excipients and raw materials used in the manufacturing of drug products
Also considered to be within the scope of Health Canada's call for review are:
- drug products that have been approved but are not yet marketed
- approved drug products with a DIN status reported as "dormant"
The request for conducting risk assessments for the potential presence of nitrosamine impurities was extended to all biological and radiopharmaceutical products for human use. This was outlined in Health Canada's letter dated December 15, 2020.
All human plasma proteins, vaccines and cell-based fermentation products are classified as biologics. They are, therefore, within the scope of the request for risk assessment.
Please refer to Health Canada's letter dated December 15, 2020, for further details.
All non-prescription products with a DIN, such as topical antiseptic products, grooming and personal hygiene products and sunscreens, are within the scope of products for assessment if they contain a chemically synthesized or semi-synthetic API. This is irrespective of the route of administration or any cosmetic properties.
Products that are not within the scope of the October 2, 2019, and December 15, 2020, letters include cosmetics (which do not have a DIN). The following categories of drug products are also excluded at this time: antimicrobial agents, veterinary products (including veterinary health products) and natural health products. Disinfectant products for use on hard surfaces are also not within the scope of products for assessment at this time.
2. Timelines for completing risk assessments (Step 1), confirmatory testing (Step 2) and changes to the market authorization (Step 3) (updated)
For drug products containing chemically synthesized and semi-synthetic APIs, the steps for actions relating to nitrosamines are expected to be completed as follows:
- Step 1: risk assessments by March 31, 2021
- Step 2: confirmatory testing by October 1, 2022
- Step 3: changes to the market authorization by August 1, 2025
For biological and radiopharmaceutical products, the steps for actions relating to nitrosamines are expected to be completed as follows:
- Step 1: risk assessments by November 30, 2021
- Step 2: confirmatory testing by November 30, 2023
- Step 3: changes to the market authorization by August 1, 2025
The following sets out Health Canada's expectations for MAHs regarding submitting and implementing necessary changes to their market authorizations effective August 1, 2025:
- For those nitrosamine impurities with an established Acceptable Intake (AI) limit published in Appendix 1 of this guidance document prior to August 1, 2025, MAHs are permitted to have a Corrective and Preventative Action (CAPA) implementation timeline of up to 3 years from August 1, 2025 (i.e., up to August 1, 2028).
- For those nitrosamine impurities with an established AI limit published in Appendix 1 on August 1, 2025 or later, MAHs are permitted to have a CAPA implementation timeline of up to 3 years from the publication date of the AI limit.
- The applicable publication dates of AI limits can be obtained from Appendix 1.
- Considering the risk profiles of nitrosamines and the possibility of an additive biological effect, the AI limits published in Appendix 1 are considered appropriate by Health Canada for lifetime and less-than-life (LTL) administration of a drug product, including during the 3 year CAPA implementation timeline. (i.e., LTL-based AI limits are not to be applied during or after 3 year CAPA implementation timeline).
It should be noted that despite the 3 year CAPA implementation timeline, it is Health Canada's expectations that MAHs implement risk mitigation measures to reduce the levels of nitrosamine impurities to below the AI limits as soon as possible.
3. Outcomes of risk assessments (Step 1) and what is provided to Health Canada
Risk assessment documentation should be retained by the MAH, unless nitrosamine impurities are detected in the API, drug product or both during confirmatory testing. Following the completion of confirmatory testing of the drug product, Health Canada must be informed if the nitrosamine impurity is detected above the established Acceptable Intake (AI) limit (refer to Appendix 1 for a listing of established AIs) for the nitrosamine impurity in question, or above the AI limit established using the Carcinogenic Potency Categorization Approach (CPCA) (refer to number 24 and Appendix 4) if an AI limit has not been established by Health Canada. The confirmatory testing results should be submitted at the same time that Health Canada is informed of the detection and the details of the risk assessment should be available upon request. Refer to the information in number 15.
For nitrosamine impurities listed in Appendix 1 that are classified as non-mutagenic, the submission of the risk assessment and confirmatory testing results is not required, and these impurities should be controlled according to ICH's Q3A and Q3B guidelines.
Please note that Health Canada may request to review the MAH's risk assessment for all products and will request this information directly from the MAH, as necessary.
Canadian importers that received terms and conditions (T&C) on their drug establishment licence (DEL) for nitrosamine testing of angiotensin II receptor blockers (also known as sartans) may provide supporting information to modify or remove the terms and conditions. They should submit the API and drug product risk assessments and testing results completed as per Steps 1 and 2 for consideration. Email to foreign.site-etranger@hc-sc.gc.ca.
MAHs may be requested by Canadian importers for a copy of the MAH's risk assessment and testing results to facilitate this request. Alternatively, MAHs may provide the requested risk assessment and related information to Health Canada on behalf of the Canadian importer. In this case, the MAH should specify on whose behalf the risk assessment and related information is being submitted.
4. Determining the priorities and order in which products should be reviewed
MAHs should use a risk-based approach to determine the order in which their drug products are reviewed. In order to prioritize the sequence in which products should be reviewed, MAHs should consider a number of factors, including the following:
- principles set out in the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Q9 guideline on quality risk management
- maximum daily dose of the drug product
- route of administration
- duration of use
- indication and consideration of special populations, such as pregnant women and children
- toxicological profile of the API
- for example, evaluating the risk of presence of nitrosamine impurities in cancer therapies in which the API is a potent mutagen could be considered lower priority and sequenced for review after higher priority APIs
- market considerations such as the availability of product for sale on the Canadian market and number of patients being treated with the drug product
- emerging international or domestic information that 1 or more nitrosamine impurities has been identified in an API (or a structurally similar API) or drug product
- the presence of structural elements in the API or conditions in the manufacturing and packaging processes for the API or drug product, which are conducive to nitrosamine formation (for example, presence of secondary or tertiary amine groups in the API)
Appendix 1 should be consulted for APIs and drug products that may contain nitrosamine impurities. Peer-reviewed literature (for example, M.K. Parr, J.F. Joseph, Journal of Pharmaceutical and Biomedical Analysis 164 (2019) 536–549) and other sources of information (for example, regulatory communications) should also be consulted for APIs and drug products known to contain nitrosamine impurities.
5. MAHs co-operating with API and drug product manufacturers to perform risk assessments
After receiving authorization to market in Canada, MAHs are responsible for the safety, efficacy and quality of their drug products and for carrying out the risk assessments. They should:
- work with API and drug product manufacturers to review their API and drug product manufacturing processes to conduct risk assessments
- take into account the API and drug product manufacturers' knowledge of the manufacturing processes, potential sources of contamination and other root causes of the formation and presence of nitrosamine impurities
API and drug product manufacturers should make available to the MAHs the information necessary for conducting the risk assessments.
If the risk of nitrosamine impurity formation has been assessed during the development phase of the API or drug product manufacturing processes, the information from the assessment can be used to support the evaluation.
6. Responsibilities of API manufacturers, excipient manufacturers, drug product manufacturers, MAHs and importers
After receiving authorization, MAHs are responsible for ensuring the ongoing safety, efficacy and quality of drug products on the Canadian market. This would include implementing an ongoing monitoring program to detect trends in quality. Such a program should be based on appropriate controls for raw materials, all processing steps, critical process parameters and critical quality attributes.
To complete risk assessments for the potential presence of nitrosamine impurities, MAHs should complete robust risk evaluations using a holistic approach. A detailed assessment of all stages of the product's life cycle should be done and would include an evaluation of the risk factors and potential root causes for the presence of nitrosamines, including those identified in number 29.
MAHs are responsible for ensuring that personnel with acceptable qualifications and expertise (for example, relevant training, knowledge and practical experience) have conducted the risk assessments. Information should be made available to the MAH by API, excipient and drug product manufacturers.
In the context of control for nitrosamine impurities, manufacturers and importers must comply with any terms and conditions specified on their DEL. This could include restrictions or additional specific testing and investigational requirements for nitrosamine impurities.
7. Inability to meet specified timelines for risk assessment (Step 1), confirmatory testing (Step 2) or changes to the market authorization (Step 3)
Given the potential risks associated with nitrosamines in drug products, MAHs should take all necessary measures to complete the 3 steps as soon as possible and within the designated timelines.
In a follow-up letter issued to MAHs of drug products containing chemically synthesized active pharmaceutical ingredients on April 14, 2021, Health Canada requested that affected MAHs indicate their status for completing Step 1, risk assessments. MAHs should provide a completed Annex 1, Annex 2 or Annex 3, as applicable, as per the instructions in the April 14, 2021, follow-up letter. If risk assessments have not been completed for all marketed, approved and dormant products or have been partially completed, Annex 3 should be completed.
MAHs unable to meet the Step 2 or 3 deadlines due to exceptional circumstances may submit a request for extension to Health Canada. This should be done as soon as possible. The request should contain relevant information, including the progress to date, the reasons for not meeting the deadline(s), the remaining work and the expected timelines for completion.
To prioritize APIs and drug products for the completion of risk assessments, confirmatory testing or changes to the market authorization, MAHs are reminded to use quality risk management principles. Consult ICH's Q9 guideline, Health Canada's good manufacturing practices (GMP) guides 0001 (for drug products) and 0104 (for APIs). Also consult the information in number 4.
Requests for extensions will be considered on a case-by-case basis. Direct such requests as follows:
- for drug products containing chemically synthesized or semi-synthetic APIs: bpsenquiries@hc-sc.gc.ca
- for biological and radiopharmaceutical products: brdd.nitrosamines.dmbr@hc-sc.gc.ca
8. Statements or declarations by manufacturers and suppliers in lieu of completing risk assessments
Statements and declarations provided by manufacturers and/or suppliers are not a substitute for an overall robust risk assessment by the MAH. While the knowledge and expertise offered by manufacturers is valuable and is encouraged to support the risk assessment process, manufacturer/supplier statements or declarations do not replace a documented risk assessment by the MAH.
9. Skipping the risk assessment step (Step 1) and proceeding directly to confirmatory testing (Step 2)
The risk assessment step (Step 1) is necessary to identify risk factors, possible root causes and the scope of nitrosamine impurities that have the potential to be formed or introduced into the API or drug product. If a risk of 1 or more nitrosamine impurities is identified, this knowledge is used to guide the development and validation of appropriate test methods required for the confirmatory testing stage (Step 2).
This knowledge may also be useful for the establishment of a suitable control strategy and changes introduced to prevent the presence of nitrosamines.
As such, it is not appropriate to proceed directly to confirmatory testing (Step 2) without completing the risk assessment step (Step 1).
10. Applying the results of a risk assessment and confirmatory testing for a drug product marketed outside Canada to a drug product authorized for sale in Canada
MAHs are responsible for ensuring that risk assessments and, if applicable, confirmatory testing are relevant to the drug product authorized for sale in Canada.
If a risk assessment and confirmatory testing have been completed for a drug product approved for use outside Canada, it may be possible to use that information for the risk assessment and confirmatory testing of the drug product authorized for sale in Canada. In this scenario, the 2 drug products must be identical (for example, composition, strength, manufacturing process, API and excipient sources, manufacturing sites).
MAHs should prepare a written justification when the risk assessment and confirmatory testing results of a foreign product will be relied upon. MAHs should be prepared to provide this justification to Health Canada upon request. This justification should be included in communications to Health Canada if nitrosamine impurities are detected in the drug product following confirmatory testing where the nitrosamine impurity is detected above the established AI limit (refer to Appendix 1) for the nitrosamine impurity in question, or above the AI limit established using the CPCA (refer to number 24 and Appendix 4) if an AI limit has not been established by Health Canada. Refer to the information in number 15.
11. Nitrosamine risk assessment applicable to a drug product brought into Canada under the Special Access Program (SAP)
Companies may need to conduct nitrosamine risk assessments for drug products not authorized for sale in Canada that are being made available in Canada through the SAP. Refer to the approaches described in Health Canada's October 2, 2019, and December 15, 2020, letters and in this document.
If the nitrosamine risk assessment or confirmatory testing results (if applicable) indicate the risk of presence of a nitrosamine impurity, notify the SAP by email at sapd-pasm@hc-sc.gc.ca.
To protect the health and safety of patients accessing unauthorized drug products, significant new information on the safety, efficacy and quality of drug products released under the SAP should be made available to practitioners and the SAP.
12. Confirmatory testing where the risk assessment concludes there is no identified risk for the presence of nitrosamines or where nitrosamine impurities can be controlled according to the ICH Q3A and Q3B guidelines (updated)
MAHs are expected to conduct a thorough, robust risk assessment for the potential formation or presence of nitrosamine impurities. In Health Canada's letters dated October 2, 2019, and December 15, 2020, Health Canada shared some potential sources of nitrosamine impurities. Refer to number 29 for more information on risk factors and potential root causes for nitrosamine impurities.
MAHs should prepare a report that includes considerations, steps, conclusions and a rationale. The report should clearly identify which nitrosamine(s) is (are) at risk of formation, if applicable. If it is concluded that a risk for the presence of nitrosamines is not identified, then confirmatory testing is not expected.
If a risk of formation or presence of nitrosamines is identified, confirmatory testing should be carried out using appropriately validated and sensitive methods (refer to number 36). Following the completion of confirmatory testing of the drug product, Health Canada must be informed if the nitrosamine impurity is detected above the AI limit established by Health Canada or above the AI limit established using the CPCA (refer to number 24 and Appendix 4) if an AI limit has not been established by Health Canada. The reporting addresses are provided in number 15.
For nitrosamine impurities that can be controlled according to the ICH Q3A and Q3B guidelines (for example, refer to number 3 and number 25), confirmatory testing is generally not expected if their risk of presence can be sufficiently mitigated based on scientific considerations that demonstrate the relevant ICH Q3A and Q3B thresholds will not be exceeded. In such cases, the scientific justification should be documented in the risk assessment and controlled by the MAH's pharmaceutical quality system.
13. Managing and submitting Step 3 changes to the market authorization relating to risk mitigation measures (updated)
Step 3 changes to the market authorization relating to risk mitigation measures should be submitted to Health Canada in a timely manner in eCTD or non-eCTD format using the Common Electronic Submission Gateway (CESG).
Step 3 risk mitigation related changes are the result of potential safety concerns and therefore require Health Canada's critical review of scientific data and subsequent authorization prior to implementation by the manufacturer. Therefore, with the exception of certain minor changes (see further details described below in this section), Step 3 risk mitigation related changes for drug products containing chemically synthesized and semi-synthetic APIs should be submitted as Level I - Supplements or Post-Drug Identification Number (DIN) Change submissions, as applicable. For biological and radiopharmaceutical products, changes should be submitted as Level I – Supplements or Level II - Notifiable Changes, or Post-Drug Identification Number (DIN) Change submissions (PDCs), as applicable. For examples of risk mitigation related changes, refer to number 20 and Appendix 2.
When filing a Supplement, Notifiable Change or Post-DIN Change submission for the market authorization, applicants should clearly indicate in the covering letter that the proposed changes are being submitted to address risk mitigation for nitrosamines (Step 3 of Health Canada's call for review to evaluate the risk of the presence of nitrosamine impurities in approved drug products).
A summary of the root cause investigations and the conclusion regarding the confirmed root causes(s) for nitrosamine presence in the drug product should be included under section 3.2.P.2.
Where a proposal is made to add individual or cumulative nitrosamine AI limits to an approved drug substance or drug product specification (based on the use of the CPCA or other approaches), MAHs should manage these changes as Level I – Supplements.
For proposed changes unrelated to Step 3 of the call for review of approved drug products (that is, changes unrelated to risk mitigation for approved drug products), changes should be managed and, where applicable, submitted according to the Post-Notice of Compliance (NOC) Quality Document and Post-Drug Identification Number (DIN) Changes Guidance Document. Refer to number 20 and Appendix 2.
For drug products containing chemically synthesized or semi-synthetic APIs, where AI limits for individual nitrosamine impurities and, where relevant, limits for cumulative levels of nitrosamine impurities, had previously been included in an approved drug substance or drug product specification based on the approval of an application for market authorization, the following changes can be managed according to Health Canada's Post-Notice of Compliance (NOC) Quality Document as Level III changes (Annual Notifications):
- tightening of individual or cumulative nitrosamine AI limits in the approved drug substance or drug product specifications
- relaxation of individual or cumulative AI limits in the approved drug substance or drug product specifications to adopt AI limits listed in Appendix 1 of this guidance document
- deletion of a test for a nitrosamine impurity from a drug substance or drug product specification, with appropriate scientific justification
In addition, the following changes can be managed according to Health Canada's Post-Notice of Compliance (NOC) Quality Document as Level III changes (Annual Notifications):
- addition of a test and acceptance criteria to a drug substance specification for a nitrosamine impurity that is based on a valid Certificate of Suitability (CEP) issued by the European Directorate of Quality of Medicines and HealthCare (EDQM)
- addition of a test and acceptance criteria to a drug substance or drug product specification for a nitrosamine impurity that has been classified as non-mutagenic in Appendix 1
Where a proposal is made to relax individual or cumulative nitrosamine AI limits already included in an approved drug substance or drug product specification (based on the use of the CPCA or other approaches) when an AI limit is not listed in Appendix 1, MAHs should manage these changes as Level I – Supplements.
Bioequivalence studies for reformulated drug products
Reformulation of drug products to include nitrosamine suppressing agents, such as antioxidants and pH modifiers, is one potential strategy to reduce or prevent the formation and presence of nitrosamine impurities. Refer to Appendix 5 for recommendations regarding bioequivalence studies for reformulated drug products.
14. Selling a drug product if changes (specifications, controls) to the market authorization (Step 3) submitted as a Supplement, Notifiable Change or Post-DIN Change submission are still under review
The ongoing marketing of a drug product depends on the impurity levels and the risk of nitrosamine impurities that are detected upon notification to Health Canada. Some of the outcomes of the Health Canada assessment may include recalls or stop sale requests until the risks are mitigated and suitable corrective and preventive actions are in place to ensure that all lots being released to market meet the acceptance criteria for each nitrosamine impurity (and multiple nitrosamines, if relevant). Both outcomes would affect the ongoing marketing of the drug product while the Supplement, Notifiable Change, or Post-DIN Change submission is being reviewed. Refer to the information in number 13.
15. Contacting Health Canada if nitrosamine impurities are detected following the completion of confirmatory testing (updated)
Following the completion of confirmatory testing of the drug product, MAHs must inform Health Canada if nitrosamine impurities are detected above the established AI limit (refer to Appendix 1) for the nitrosamine impurity in question, or above the AI limit established using the CPCA (refer to number 24 and Appendix 4) if an AI limit has not been established by Health Canada. The confirmatory testing results should accompany the notification to Health Canada by the MAHs.
Health Canada recognizes the challenges faced by MAHs to decrease levels of nitrosamine impurities in their drug products while maintaining drug supply to Canadians. To minimize the impacts on drug supply within the Canadian market, MAHs are requested to engage Health Canada prior to taking any market action for a drug product due to a nitrosamine impurity issue.
Communications should be directed as follows:
| Location of firm | Reporting address |
|---|---|
| New Brunswick, Newfoundland and Labrador, Nova Scotia, Prince Edward Island, Québec | Health Product Compliance Unit East |
| Ontario | Health Product Compliance Unit Central |
| Manitoba, Saskatchewan, Alberta, British Columbia, Yukon, Northwest Territories, Nunavut | Health Product Compliance Unit West |
If nitrosamines are not detected during confirmatory testing (for example, less than the appropriate limit of detection of the validated test method) or are detected below the established AI limit for the nitrosamine impurity in question or the AI limit established using the CPCA (refer to number 24 and Appendix 4) if an AI limit has not been established by Health Canada, MAHs do not need to communicate this information to Health Canada. However, they should keep the risk assessment, analytical testing results and analytical method validation documentation on hand in case Health Canada requests them. Refer to number 13 for information on communicating changes to market authorization to Health Canada.
16. When information necessary to complete risk assessments is not provided by the API or drug product manufacturer
MAHs are responsible for ensuring the ongoing safety, efficacy and quality of products on the Canadian market. When manufacturers do not provide information that is essential for MAHs to complete the risk assessment due to confidentiality or other reasons, MAHs may engage a third party (such as a consultant) to work directly with the manufacturer to complete the risk assessment.
The third-party approach may also be appropriate when the MAH:
- has all of the required information to conduct the risk assessment from the manufacturer but
- does not have staff with the necessary qualifications (for example, relevant training and practical experience) to conduct the risk assessment
For additional guidance on outsourced activities, consult the:
Alternatively, MAHs should consider delegating the risk assessment to the API and drug product manufacturers. In this scenario, MAHs would continue to be responsible for ensuring the safety, efficacy and quality of their drug products.
MAHs should ensure through internal or third-party audit that:
- risk assessments have been conducted by personnel with acceptable qualifications (relevant training and practical experience)
- manufacturers have considered all possible risk factors and potential root causes of nitrosamine impurities (including those in the December 15, 2020, letter concerning biological and radiopharmaceutical products, and those identified in number 29)
17. Additional expectations of MAHs if nitrosamine impurities are detected in the API and/or drug product (updated)
Where 1 or more nitrosamine impurities are detected following the completion of confirmatory testing (for multiple nitrosamines, refer to number 27), in addition to notifying Health Canada (refer to number 15 for detailed guidance on when to notify), MAHs should have completed or be completing as necessary:
- a health risk assessment posed by the presence of the nitrosamine(s) along with intentions related to any actions, as necessary, for the batches on the Canadian market
- where product recalls are warranted, consult the Drugs and Natural Health Products Recall Guide (GUI-0039) for procedures
- an assessment to determine if the product is considered to be medically necessary or medically important and if any disruption to product supply is expected should market action be taken
- a detailed investigation report assessing all possible root causes of the detected nitrosamine impurity (or impurities) and describing corrective and preventive actions
- perform investigations in accordance with written procedures
- evaluate all potential changes to facilities, materials, equipment and/or process intended to reduce the levels of the nitrosamine impurities through a formal change control system
- a risk mitigation plan including the establishment of a suitable control strategy for detected nitrosamine(s) to ensure that, moving forward, nitrosamine impurity levels will be consistently below the Acceptable Intake (AI) limit at the end of the retest period for the API or the shelf-life for the drug product. Refer to Appendix 1 for a list of established AI limits. These AI limits are considered qualified for nitrosamine impurities in the drug product at the end of shelf life. In some cases, tighter limits in the API specifications and in the drug product release specifications may be warranted to ensure that the drug product shelf life specification will be met.
MAHs are reminded to submit changes to the market authorization as per Step 3 of the October 2, 2019 letter. Refer to number 13 on how changes should be submitted.
Health Canada may use such notifications to request additional actions and/or information. For example, the origin of nitrosamine impurities may be attributed to the type of process chemistry used and the risk mitigation plan may necessitate the establishment of a control strategy by manufacturers for each detected nitrosamine impurity according to ICH's M7 guideline.
We may request additional actions by other MAHs of the same products to mitigate any risks identified and protect people's health and safety if necessary.
For more information on the establishment of specifications and controls, refer to number 34 and number 35, respectively.
18. Assessing progress with the request to review the risk of presence of nitrosamine impurities
On April 14, 2021, Health Canada issued a follow-up letter to the October 2, 2019, letter. In the letter, we asked MAHs with drug products containing chemically synthesized and semi-synthetic APIs to provide their progress towards completing Step 1 (risk assessments).
Health Canada may also:
- verify progress, adequacy and robustness of the risk assessments, confirmatory testing results and any other supporting documents related to this request during GMP inspections, proactive risk management projects and compliance verification upon receipt of a complaint and/or notification or
- request information at such time as changes are made to either the existing market authorization for a product or for the drug establishment licence
We appreciate the significance of this request. We will continue to engage with stakeholders to consider all options to address the potential risks associated with nitrosamines.
19. Approach for drug products that are planned for submission or are already filed with Health Canada
Whenever possible for APIs and drug products that are under development, the formation or introduction of nitrosamine impurities should be avoided at the outset.
If the formation or introduction of nitrosamine impurities is unavoidable, manufacturing processes should demonstrate process capability to routinely reduce the levels of nitrosamine impurities below the AI limit. A control strategy, based on product and process understanding, should be established for each nitrosamine impurity present in the API and/or drug product.
For drug products that are planned for submission or have already been submitted, MAHs and applicants should proactively undertake a risk assessment for the potential presence of nitrosamine impurities in the drug product (if this has not already been undertaken) using the considerations and steps provided for approved products in Health Canada's communications. For planned submissions, the relevant sections of the Common Technical Document (CTD) in the drug application should include information on these risk assessments.
A summary and discussion of the risk assessment for nitrosamine impurities in the drug product should be placed in section 3.2.P.2 of the CTD. This summary is expected to include sufficient detail to allow Health Canada to assess the adequacy and robustness of the risk assessment. Expectations for the content of the summary and discussion of risk assessments are found under number 20.
Confirmatory testing results and the updated control strategy (where warranted) should also be included in the drug application (for example, under sections 3.2.S.2, 3.2.S.4, 3.2.S.7, 3.2.P.3, 3.2.P.4, 3.2.P.5, 3.2.P.8).
For submitted applications currently under review, MAHs and applicants may be asked to provide the risk assessment and confirmatory testing results as part of the assessment procedure. For further information, refer to number 20.
20. Risk assessments for the potential presence of nitrosamine impurities as part of the expected content for new submissions (updated)
Risk assessments for the potential presence of nitrosamine impurities should be conducted routinely during API and drug product development. The outcome of the risk assessment for nitrosamine impurities in the drug product and the justification for the proposed control strategy for nitrosamine impurities should be made available for assessment in New Drug Submissions (NDSs), Abbreviated New Drug Submissions (ANDSs), applications for a Drug Identification Number for a Pharmaceutical Product (DINAs) (with Chemistry & Manufacturing (C&M) data), applications for a Drug Identification Number for a Biological Product (DINBs), Supplements, Notifiable Changes and Post-DIN Changes submissions (refer to number 13). For more information on mutagenic impurity considerations and quality risk management principles, consult the following:
- Good Manufacturing Practices Guide for Drug Products (GUI-0001)
- Good Manufacturing Practices Guidelines (GMP) for Active Pharmaceutical Ingredients (GUI-0104)
- ICH's M7 guideline (PDF format)
- ICH's Q9 guideline (PDF format)
All NDSs, ANDSs, DINAs (with C&M data), DINBs, and as detailed below, certain Supplements, Notifiable Changes and Post-DIN Change submissions (for quality changes that may increase the risk of presence of nitrosamine impurities relative to the approved drug product) should include a summary and discussion of the risk assessment for the potential presence of nitrosamine impurities in the drug product.
For Supplements, Notifiable Changes and Post-DIN Change submissions where the quality changes do not increase the risk of presence of nitrosamine impurities relative to the approved drug product (for example, the addition of a proportionally-formulated strength), a summary and discussion of the risk assessment for the potential presence of nitrosamine impurities in the drug product is not expected to be provided in the drug submission. This approach is taken as the potential nitrosamine-related risks for the approved drug product are expected to be addressed and managed under Health Canada's call for review. In such cases, a brief justification for the conclusion that there will be no increased risk for nitrosamine presence relative to the approved drug product as a result of the proposed changes should be provided, for example, in the Notes to Reviewer or in Sections P.2 or P.5 of the drug submission.
This is required as follows:
- as of April 1, 2021, for NDSs, ANDSs, and Supplements for pharmaceutical products containing chemically synthesized and semi-synthetic APIs
- as of April 1, 2021, for DINAs (with C&M data) including Post-DIN Change Submissions for quality changes for pharmaceutical products containing chemically synthesized and semi-synthetic APIs
- as of November 30, 2021, for NDSs, Supplements and Notifiable Changes for biological and radiopharmaceutical products
- as of November 30, 2021, for DINBs and Post-DIN Change (PDC) submissions for biological and radiopharmaceutical products
The summary and discussion of the risk assessment for the drug product is expected to include sufficient detail to allow Health Canada to assess the adequacy and robustness of the risk assessment. It should include a discussion of the risk factors and potential root causes considered in relation to specific knowledge of the drug product and its components (including the API). Checklists lacking sufficient discussion and detail should be avoided. The summary and discussion should include the following:
- identification of any third parties (for example, suppliers, manufacturers, consultants) who have been authorized to perform the risk assessment on behalf of the applicant
- identification of intrinsic and extrinsic risk factors related to formation or introduction of nitrosamine impurities originating from all drug product components as well as quality/compliance considerations
- identification of those nitrosamines potentially formed and/or introduced
- information on the established process and/or analytical controls and how they may mitigate risk
- supporting scientific data (for example, confirmatory testing results) and calculations
- an overall conclusion on the risk of presence of nitrosamines in the drug product together with an appropriate scientific rationale/justification
For certain Supplements, Notifiable Changes and Post-DIN Change submissions (for quality changes that may increase the risk of presence of nitrosamine impurities relative to the approved drug product), the summary and discussion of the risk assessment need only address the impact of the proposed change(s) on nitrosamine impurities relative to the approved drug product. Examples of changes that may impact the potential presence of nitrosamine impurities relative to an approved drug product include, but are not limited to, changes in drug substance or drug product manufacturing processes, changes to the drug product composition (API, excipients), introduction of a new dosage form, and changes to the container closure system.
Refer to Appendix 2 for additional guidance with respect to nitrosamine impurities and risk assessments for Post NOC Changes of new drug products containing chemically synthesized and semi-synthetic APIs.
For Clinical Trial Applications (CTAs) (as described in section 9.1 of ICH's M7 guideline):
- For Phase 1 clinical trials of 14 days or less, include a description of efforts to mitigate risks of mutagenic impurities focused on Class 1 and Class 2 impurities and those in the cohort of concern (for example, nitrosamine impurities).
- For Phase 1 clinical trials greater than 14 days and for Phase 2 and 3 clinical trials, also include Class 3 impurities that require analytical controls.
Failure to include this information could result in requests for additional information, delays in the review process, and potentially the issuance of negative decisions.
21. Controls for nitrosamines in APIs purchased for compounding
The Policy on Manufacturing and Compounding Drug Products in Canada (POL-0051) provides a policy framework to help distinguish between compounding and manufacturing activities of drug products in Canada. In Canada, the compounding of drugs is done mainly by pharmacists as an integral part of their profession. It's regulated by the respective regulatory authorities in each province/territory.
This policy indicates that compounded products should be either:
- produced from an authorized API used in an authorized drug product for use in Canada or
- listed in a recognized Pharmacopoeia (for example, USP/NF, Ph. Eur., Ph. Int., BP, Codex - Schedule B, Food and Drugs Act)
Health Canada recommends, therefore, that principles outlined in the October 2, 2019, letter and in this guidance should be considered when purchasing APIs and producing compounded products.
Health care professionals and compounding firms are encouraged to access the nitrosamines webpage to stay informed on product recalls due to the presence of nitrosamines. This page is updated regularly and also includes general information on nitrosamine impurities, and what Health Canada is doing to address the issues.
Communications
22. Engaging stakeholders and ensuring ongoing communication with industry
Health Canada is committed to sharing information with stakeholders and maintaining transparency as we continue to analyze and better understand this evolving, global situation.
To date, we have shared information openly with stakeholders, including information on the potential sources of nitrosamine impurities, root causes, risk factors and new findings. We hosted stakeholder sessions in January 2020, February 2021 and October 2021, and may host more sessions in the future if necessary.
We also established a dedicated webpage on nitrosamine impurities in medications. The webpage includes the following:
- summaries of drug products that have been affected or recalled due to the presence of nitrosamines
- analytical testing results of several products for levels of nitrosamine impurities
Discussions are ongoing to determine the most appropriate and effective methods to continue to engage stakeholders as new information becomes available to ensure a coordinated and consistent approach in dealing with this complex issue.
23. Health Canada works with global regulators relating to issues associated with nitrosamine impurities in drug products
Health Canada regularly collaborates with international regulatory partners, including those in Europe, the United States, Japan, Switzerland, Singapore, Australia and Brazil, as well as the World Health Organization. Through collaboration, we hope to increase the understanding of the issues associated with nitrosamine impurities, align requirements and actions as appropriate, and share information under the terms of our confidentiality agreements.
Discussions amongst the consortium of regulatory authorities continued, with the "Nitrosamines International Strategic Group" (NISG), which was formed in 2018. The NISG began meeting regularly with a focus on knowledge sharing on market and other regulatory actions. Due to a mutual interest of the participating regulatory agencies of the NISG to have greater in-depth discussions on technical issues and scientific developments, a sub-group of the same regulators, the "Nitrosamines International Technical Working Group" (NITWG), was established in late 2020.
When determining appropriate regulatory measures to address the presence of nitrosamine impurities that exceed the AI limit in human drug products, individual jurisdictions must determine timelines and actions that will best protect patient safety and work within the relevant regulatory framework.
Safety
24. AI limits for nitrosamine impurities that Health Canada considers acceptable
AI limits have been derived for several nitrosamines (Appendix 1). These AI limits are considered appropriate for all routes of administration and should be applied to the maximum daily dose (MDD) of the drug product.
AI limits can be established using several approaches.
In cases where there is reliable compound-specific data for a nitrosamine impurity, MAHs and applicants may:
Establish an AI limit based on reliable compound-specific data
- Linearly extrapolate from the dose giving a 50% tumour incidence (TD50) to a 1 in 105 excess cancer risk, using the most relevant TD50 value from a sufficiently robust carcinogenicity study (refer to ICH's M7 Addendum for guidance on selecting an appropriate carcinogenicity study).
- Provide a negative, GLP-compliant, enhanced Ames test using the enhanced Ames test conditions described in Appendix 3 to justify a limit of 1.5 µg/day.
- Provide negative in vivo mutagenicity data (for example, a negative in vivo mutagenicity assay conducted per the Organisation for Economic Co-operation and Development (OECD)'s Test Guideline No. 488 "Transgenic Rodent Somatic and Germ Cell Gene Mutation Assays") to justify controlling a nitrosamine impurity per the recommendations in ICH's Q3A and Q3B guidelines.
In cases where there is insufficient reliable compound-specific data available for a nitrosamine impurity, MAHs and applicants may:
Establish an AI limit based on a structure-activity relationship (SAR) assessment and read-across to a surrogate with sufficient compound-specific data
To justify an appropriate surrogate for read-across, the SAR assessment should take into consideration structural similarity (both overall and at the local site of activation), similarity of physicochemical characteristics, steric and electronic factors impacting reactivity and metabolic similarity (for example, metabolic pathway, stability/reactivity of metabolites).
If an appropriate surrogate for read-across is identified, to calculate an AI limit, the TD50 should be derived from a sufficiently robust carcinogenicity study. Parameters to consider include adequate description of the study design and appropriate histopathological analysis, number of dose groups (i.e., single-dose studies are not considered appropriate), number of animals per dose group, duration of exposure, route of administration, observed dose-response relationship. Refer to ICH's M7 Addendum for guidance on selecting an appropriate carcinogenicity study.
Alternatively, in cases where an acceptable surrogate for read-across may be controlled to a limit as per the recommendations in ICH's Q3A and Q3B guidelines (when indicated in Appendix 1), this limit can be applied to the nitrosamine impurity.
Consistent with international regulatory practices, Health Canada will continue to use, and expect applicants and MAHs to use, mass-based calculations (rather than molar-based) to derive AI limits for nitrosamine impurities when a surrogate is selected for read-across.
Establish an AI limit using the Carcinogenic Potency Categorization Approach (CPCA)
The Carcinogenicity Potency Categorization Approach (CPCA) is an approach for assigning a nitrosamine to a predicted carcinogenic potency category.
A total of five carcinogenic potency categories are available, each with a corresponding AI limit that ranges from 18 ng/day to 1500 ng/day.
A nitrosamine is assigned to a predicted carcinogenic potency category based on an assessment of alpha hydrogen atoms and activating or deactivating structural features present in the nitrosamine. Refer to Appendix 4 for a description of the approach which also includes case examples to illustrate application of the CPCA.
MAHs and applicants should also refer to the following items for more information:
- number 27 on the presence of multiple nitrosamines
- number 28 on applying a less-than-lifetime limit
- number 31 on which nitrosamines should be included in risk assessments and confirmatory testing
25. AI limits for nitrosamine impurities in drug products that fall within the scope of ICH's S9 guideline or where the API is genotoxic
If a nitrosamine impurity is identified in a pharmaceutical, biological or radiopharmaceutical product that is intended for advanced cancer indications (defined in the scope of ICH's S9 guideline), the impurity can be controlled per the recommendations in ICH's S9 questions-and-answers document.
If a nitrosamine impurity is identified in a drug product where the API is genotoxic at therapeutic concentrations, the impurity can be controlled at limits for non-mutagenic impurities. Refer to ICH's Q3A and Q3B guidelines.
26. Communicating if AI limits are revised in the future
Health Canada continues to work with international regulatory agencies to determine AI limits for nitrosamine impurities. We will communicate any changes to the AI limits for nitrosamine impurities to MAHs and applicants in a timely manner.
Interim AI limits were originally communicated to MAHs for 5 nitrosamine impurities in angiotensin II receptor blockers (also known as "sartans"). These were in place until September 30, 2020, and will not be reduced to a lower level.
27. Acceptable Intake limit if multiple nitrosamines are detected in an API or a drug product
If an API or drug product has the risk of containing more than 1 actual or potential nitrosamine impurity, total (cumulative) daily exposure should be limited to the nitrosamine with the most conservative AI limit at the maximum daily dose of the drug product.
Examples:
- If a drug product contains both NDMA and NMBA, the total/cumulative daily exposure of the 2 nitrosamines should be limited to 96.0 ng/day.
- If a drug product contains both NDMA and NDEA, the total/cumulative daily exposure of the 2 nitrosamines should be limited to 26.5 ng/day.
If an applicant or MAH proposes to control multiple nitrosamine impurities in an API or drug product using an alternative methodology, Health Canada will assess the acceptability of the approach on a case-by-case basis. Any proposed alternative methodology should ensure that excess cancer risk does not exceed 1 in 100,000.
For nitrosamine impurities that are considered non-mutagenic, the recommendations provided in ICH's Q3A and Q3B guidelines apply and such impurities do not need to be included in a limit for total nitrosamines.
28. Application of a less-than-lifetime (LTL) limit by considering the principles in ICH's M7 guideline if a nitrosamine impurity is present in a drug product that is administered for less than a lifetime
Considering the risk profiles of nitrosamines and the possibility of an additive biological effect, the AI limits outlined in Appendix 1 are considered appropriate for lifetime and LTL administration of a drug product.
If a nitrosamine impurity cannot be controlled at the AI limit, Health Canada may consider an interim limit higher than the AI limit. We will do so on a case-by-case basis and only in exceptional circumstances (for example, to avoid a drug shortage of a drug product that is considered medically necessary or medically important).
Where an applicant or MAH proposes an interim limit higher than the AI limit for a nitrosamine impurity, Health Canada will consider:
- the medical necessity or medical importance of the drug product
- levels of impurity observed in representative batches
- other risk management considerations (for example, the availability of alternative medications on the Canadian market)
We will consider an interim limit higher than the AI limit for a nitrosamine impurity as a transitory measure only, until appropriate changes to reduce the level of the nitrosamine impurity to at or below the AI limit have been implemented.
For nitrosamine impurities that are considered non-mutagenic, the recommendations provided in ICH's Q3A and Q3B guidelines apply and such impurities do not need to be included in a limit for total nitrosamines.
Quality
29. Risk factors and potential root causes to be considered for the presence of nitrosamine impurities in human pharmaceuticals when performing a risk assessment (updated)
Knowledge of risk factors and potential root causes for nitrosamine impurities continues to evolve. Applicants and MAHs should stay up-to-date on risk factors and potential root causes that Health Canada and other regulators have identified in their guidances and peer-reviewed publications. The collaboration of the quality technical experts of the international regulatory partners of the NITWG has led to the development and publication of the paper "Regulatory Experiences with Root Causes and Risk Factors for Nitrosamine Impurities in Pharmaceuticals" (Horne et al. J. Pharm. Sci. 2023, 112, 1166-1182). This publication is designed to share current information and experiences from a quality perspective on root causes, risk factors, and risk mitigation measures relating to nitrosamine impurities in pharmaceuticals for human use.
Inadequate process design and/or process controls, as well as gaps in quality and compliance oversight, may contribute to the presence of nitrosamine impurities in APIs and drug products above AI limits. Applicants and MAHs should consider both intrinsic and extrinsic factors when conducting risk assessments for nitrosamine impurities.
Potential and confirmed root causes and risk factors for the presence of nitrosamines in drug products include the following:
- Nitrosation of a secondary or tertiary amine during API or drug product manufacturing, with insufficient downstream purge of the nitrosamine formed, and/or during API or drug product storage (common nitrosation conditions involve the combination of amines and nitrite ion under acidic conditions)
- Sources of amines include APIs, API intermediates, starting materials, reagents, solvents, catalysts, reaction by-products and degradation products. Certain non-medicinal ingredients may also contain amines as part of their structure. Amines leading to stable nitrosamines include secondary amines and tertiary amines. Quaternary ammonium salts are also potential precursors to nitrosamines. Primary and tertiary amines may contain secondary amines as impurities. Amines may be present as impurities in amides or formed through the degradation of amides (for example, via hydrolysis). Tertiary amines may be nitrosated by a dealkylative pathway to produce one or more secondary amines, which may subsequently undergo nitrosation to produce multiple nitrosamines.
- Nitrosating agent precursors and sources include:
- nitrite ion intentionally used in a manufacturing process (for example, as used in diazotization chemistry or as a reducing agent for azide ion)
- nitrite present as an impurity in reagents (for example, sodium azide), common non-medicinal ingredients (for example, microcrystalline cellulose, magnesium stearate)
- nitrogen oxides (for example, NO, N2O3)
- nitric acid
- nitrosyl halides
- alkyl nitrites and nitro compounds (for example, nitromethane)
- potable and/or purified water containing nitrite
- Using a nitrosamine as a starting material or synthetic intermediate, with incomplete conversion of the nitrosamine and/or insufficient downstream purge
- Reaction of nitrite ion and an amine under process conditions with pH >7 under catalysis by a carbonyl compound, with insufficient downstream purge
- Oxidation of a hydrazine functional group in an API, starting material, intermediate or a reagent to produce a nitrosamine, with insufficient downstream purge
- Using certain materials in container closure components which may provide a source of nitrosating agents or amines, such as:
- nitrocellulose, found in certain lidding foils used for blister packaging
- certain types of vulcanisation accelerators (for example, dithiocarbamate, thiourea, thiruams) which are used in rubber manufacturing
- Using recycled materials (for example, solvents, reagents, catalysts) contaminated with nitrosamines and/or nitrosamine precursors
- Cross-contamination of materials with nitrosamines/nitrosamine precursors in multi-product facilities (for example, through the use of shared equipment)
- Poor operation of a process step (for example, during liquid-liquid phase separations), which is intended to purge nitrosamines
- Using certain manufacturing operations that could facilitate contact between nitrosamine precursors (for example, wet granulation) or introduce nitrosating agents/precursors (for example, nitrogen oxides during fluid bed drying)
30. Components of drug products to consider in risk assessments
All components of the drug product should be considered as potential sources of nitrosamine impurities, or their precursor nitrosating agents and amines, in the context of the designated process and storage conditions. For example, some excipients may contain residual levels of nitrite (see Wu, Y. et al. AAPS PharmSciTech 2011, 12(4), 1246-1263 and Boetzel et al. J. Pharm. Sci. 2023, 112, 1615-1624) or reactive amines as part of their molecular structure. Under certain manufacturing process or storage conditions, this may lead to the formation of nitrosamine impurities.
Refer to number 29 for more information on risk factors and potential root causes to take into consideration.
31. Nitrosamine impurities to consider in the Step 1 risk assessment and Step 2 confirmatory testing
Each API and drug product manufacturing process is unique. Thus, the nitrosamines listed in Appendix 1 of this guidance are not exhaustive and do not represent all nitrosamines potentially present in APIs and drug products. Conversely, the nitrosamines listed in Appendix 1 may not be potential impurities in all APIs and drug products.
MAHs and applicants should ensure that the risk assessments consider and identify the possibility of any nitrosamine impurity that may be formed or introduced. All nitrosamines that have been determined to be potentially formed or introduced should be included within the program for confirmatory testing (Step 2).
For nitrosamines not included Appendix 1, MAHs and applicants should refer to number 24 for guidance on how to establish an AI limit.
32. Testing methodologies provided by Health Canada
Several regulators, including Health Canada, Europe's network of Official Medicines Control Laboratories (OMCLs) and the U.S. Food and Drug Administration (FDA), have published and shared testing methodologies. These methods may be used, although there is no requirement to do so.
In all cases, companies should use appropriately sensitive, validated analytical methods and conduct the testing at a GMP-compliant facility. If other methodologies are used, there is no need to verify the method with Health Canada prior to use.
Analytical methods should be quantitative in nature (as opposed to limit-based tests) and should be fully validated before confirmatory testing begins. If limit-based tests are used, ensure that the appropriate scientific justification is provided in the risk assessment documentation. For example:
- demonstration that the limit test is valid at or lower than the AI limit
- supporting evidence that indicates there is no increase in the concentration of nitrosamine impurities over time
Unless otherwise justified, method validation should be performed using the drug product that is authorized for use in Canada.
Where multiple strengths of a drug product exist and the validation is to cover multiple strengths, the justification for the choice of product strength used for validation should be described in the validation protocol.
33. Validating the limit of quantitation (LOQ) for nitrosamine impurity analytical procedures (updated)
The LOQ for analytical procedures that are intended for quantitation of individual nitrosamine impurities in APIs and drug products should be equal to or less than the established AI limit (Appendix 1), the AI limit established using the CPCA (refer to number 24 and Appendix 4) if an AI limit has not been established by Health Canada, or the relevant ICH Q3A or Q3B reporting threshold for a nitrosamine impurity that has been classified as non-mutagenic in Appendix 1.
Where instrument sensitivity challenges exist to achieve the desired LOQ, some exceptions may be acceptable with appropriate scientific justification (for example, for drug products with a very high maximum daily dosage and nitrosamine impurities with very low AI limits).
Analytical procedures should be validated with a LOQ which is less than or equal to 10% of the acceptable limit for an individual nitrosamine, if a proposal to not routinely test for the nitrosamine in the drug product specification is anticipated (refer to number 34).
Analytical procedures should be validated with a LOQ which is less than or equal to 30% of the acceptable limit for an individual nitrosamine, if a proposal for periodic (skip) testing is anticipated.
34. Including routine testing for nitrosamine impurities in the API and/or drug product specification (updated)
The API specification should include a test and acceptance criterion for each nitrosamine impurity when:
- the risk for nitrosamine presence is considered to be high and/or
- the concentration of any nitrosamine is found to be at significant levels (for example, greater than 30% of the AI limit) during confirmatory testing
Examples where the risk of presence for nitrosamines in the API is considered high:
- potential for nitrosamine formation on storage is identified
- presence of nitrosamine precursor functional groups in the API structure
- late-stage formation/introduction of a nitrosamine impurity in the API manufacturing process
Where multiple nitrosamines are detected in an API, a cumulative limit should also be included in the specification using one of the approaches outlined in number 27.
Routine testing for nitrosamine impurities should be included in the drug product specification when:
- the potential for nitrosamine introduction during drug product manufacturing, packaging and storage is identified and/or
- a nitrosamine impurity is detected in the drug product during confirmatory testing and the root cause is unknown
If the root cause for the presence of a nitrosamine impurity in the drug product is well understood, then alternative control strategies may be acceptable when scientifically justified.
A test and acceptance criteria for both release and shelf life specifications should be included. Refer to Appendix 1 for a list of established AI limits. These AI limits are considered qualified for nitrosamine impurities in the drug product at the end of shelf life. In some cases, tighter limits in the API specifications and in the drug product release specifications may be warranted to ensure that the drug product shelf life specification will be met.
Where multiple nitrosamines are detected, control for total nitrosamines using one of the approaches outlined in number 27 should be included in the specification. Alternatively, control limits expressed on an individual impurity basis (for example, a limit for each nitrosamine set at a percentage of its AI limit such that the sum of the % AI limits for each specified nitrosamine does not exceed 100%) may be proposed with appropriate justification. Other approaches for establishing a suitable specification when multiple nitrosamines are concerned may be acceptable if appropriately justified.
The presence of one or more nitrosamines at <10% of their individual AI limits in a drug product constitutes a negligible toxicological risk; if the root cause for the presence of such nitrosamine impurities is understood and appropriate controls have been established to ensure such impurities will consistently be <10% of their individual AI limits, then such impurities do not need to be specified in the drug product specifications. Nitrosamines present below 10% of their respective AI limit do not need to be factored into the calculation of limits for total nitrosamines.
Where nitrosamine impurities can be controlled according to ICH Q3A and Q3B guidelines (refer to number 3 and number 25) and the risk of presence can be sufficiently mitigated based on a scientific justification that demonstrates the relevant ICH Q3A and Q3B thresholds will not be exceeded, then additional controls specific for nitrosamines in the API and drug product specification are not expected.
MAHs should test all new lots of drug product for nitrosamines and only release lots that meet the acceptance criteria for individual (and multiple nitrosamines, if relevant). Continue routine testing of all drug product lots until the root cause is identified and alternative controls/risk mitigation measures (such as process controls, raw material specifications) have been implemented. Ensure that nitrosamine impurities will be routinely below the AI limit in the future.
35. Potential control options for nitrosamine impurities in the API
Control options for nitrosamine impurities include:
- routine testing in the API (ICH's M7 Option 1)
- control in upstream intermediate specifications at the AI limit (ICH M7 option 2) (when the route cause, or causes, of nitrosamine presence have been established unequivocally)
- control in upstream intermediate specifications at acceptance criteria that exceed the AI limit (ICH's M7 Option 3) (when the root cause, or causes, have been established unequivocally and justification of the proposed limit is supported by demonstrated process capability (for example, spike and purge studies))
Proposals for an ICH M7 Option 4 control strategy for nitrosamine impurities in a new market authorization application will be evaluated on a case-by-case basis. An Option 4 control strategy proposal may not be appropriate when the concentration of any nitrosamine impurity in an API is greater than 30% of the AI limit. However, such a strategy may be acceptable when process understanding has been demonstrated by fate-purge studies, identification of process parameters that impact nitrosamine impurity levels and when supported by appropriate analytical data. Predicted purge factor calculations should be supported by appropriate analytical data.
This information should be provided along with copies of analytical procedures and method validation reports in the new market authorization application.
Refer also to number 34 for information on routine testing for nitrosamine impurities in the API and/or drug product specification.
36. Confirmatory testing expectations (Step 2)
During confirmatory testing, MAHs should test the drug product to determine the levels of nitrosamine impurities.
Testing the API is also recommended if the risk assessment indicated that the API is a potential source of nitrosamine impurities in the drug product. The API testing results may be used to support root cause investigations and the development of a justified control strategy for nitrosamine impurities in the API.
If a drug product is available in multiple strengths of the same dosage form with the same risk factors applicable to each, then testing could be rationalized by testing only the worst-case scenario strength. The worst-case approach should be justified by the MAH on a case-by-case basis. The justification should be documented in the risk assessment in the MAH's pharmaceutical quality system.
If, despite extensive efforts, it becomes apparent that a nitrosamine impurity cannot be synthesized or isolated and purified, then this could be an indication that the nitrosamine either does not exist, is unstable, or that there is no risk of it being formed. In such cases, it may not be necessary to conduct confirmatory testing. This should be justified thoroughly on a case-by-case basis according to appropriate scientific principles. A scientific justification including experimental data which summarizes the efforts made to synthesize and/or isolate and purify the impurity should be included in the summary and discussion of the risk assessment in the regulatory submission and documented in the MAH's pharmaceutical quality system.
37. Analytical laboratories conducting nitrosamine testing and listing on the DEL
The analytical lab used for nitrosamine confirmatory testing (Step 2) does not have to be listed on the DEL at this time. However, in all cases, the confirmatory testing must be conducted at a GMP-compliant facility. A foreign analytical lab, if used for conducting the nitrosamine confirmatory testing, must either:
- have been deemed GMP-compliant by Health Canada or
- have a valid GMP inspection by a competent or qualified regulatory authority demonstrating compliance with current GMP standards
Ethical drugs are those that do not require a prescription, but are generally prescribed by a medical practitioner as professional use products (for example, hemodialysis solutions, nitroglycerine). For testing ethical and over-the-counter drugs, if no inspection reports by regulatory/qualified authorities are available, a corporate or consultant audit report to demonstrate GMP compliance is acceptable.
For more information about acceptable GMP evidence and regulatory requirements, refer to the following guidance:
However, analytical labs must be listed on the applicable annex of the DEL if they are conducting:
- nitrosamine testing used to release APIs and drug products for the Canadian market or
- tests that are part of the API or drug product specification
- includes testing imposed through the sartan terms and conditions
For guidance on Health Canada's expectations for testing facilities or for other related questions, please send an email to foreign.site-etranger@hc-sc.gc.ca.
38. Number and types of drug product batches as part of confirmatory testing for marketed products and new market applications
For marketed products, the number of batches to be tested should be commensurate with the risk. Examples of high risk include:
- late-stage formation/introduction of a nitrosamine impurity in a manufacturing process
- presence of nitrosamine precursor functional groups in the API
- potential for nitrosamine formation on storage
MAHs and manufacturers should test a representative number of batches of the drug product as appropriate based on the risk assessment (for example, batches that are representative of sources of components, manufacturing processes/sites, manufacturing dates).
If the root cause for the nitrosamine presence has been identified and scientifically demonstrated, and impurity levels are expected to be consistent from batch-to-batch (for example, as demonstrated by spike-purge studies), testing should be conducted on 10% of annual batches, or 3 per year, whichever is highest. Testing should include both newly produced batches as well as retained samples of batches still within the expiry date. If fewer than 3 batches are manufactured annually, then all batches within the expiry date should be tested.
Testing plans or protocols (for example, a protocol for the number and type of batches to be tested) do not need to be submitted to Health Canada for assessment and approval before initiating confirmatory testing.
If nitrosamine impurities are detected at significant levels (approaching, at or above AI limits), additional batches of the drug product on the Canadian market and within the expiry date should undergo confirmatory testing. In such cases, MAHs may be requested to test all lots on the Canadian market that are within the expiry date.
For NDSs, ANDSs, DINAs (with C&M data), DINBs, Supplements Notifiable Changes and Post-DIN Change (PDC) submissions(for quality changes that may impact the potential presence of nitrosamines in the drug substance or drug product, refer to number 13), at least 6 pilot or 3 commercial-scale batches should undergo confirmatory testing where a risk of nitrosamines has been identified. A higher number of batch results should be submitted for assessment where the risk of nitrosamine presence is high. Examples include:
- the late-stage formation/introduction of a nitrosamine impurity
- nitrosamine precursor functional groups in the API
- stability concerns exist for nitrosamine formation over the retest period/shelf life
Testing of stability batches for a nitrosamine impurity should be conducted where:
- a risk has been identified that nitrosamine levels could increase in the API or drug product over time or
- the potential for increases over time is unclear
Where applicable, testing for the nitrosamine impurity for a minimum of 6 months of accelerated and long-term stability data in the proposed container closure system(s) should be provided in the drug application.
Appendices
Appendix 1: Established Acceptable Intake (AI) limits for N-nitrosamine impurities
- See separate Appendix 1.
Appendix 2: Guidance with respect to nitrosamine impurities and risk assessments for Post NOC Changes of new drug products containing chemically synthesized and semi-synthetic APIs (updated)

Appendix 2 - Text description
An image of a decision tree is providing advice to Market Authorization Holders about regulatory filing requirements related to nitrosamines. It describes some types of changes which may impact the possibility of nitrosamine formation when a change is being proposed. Are changes to a market authorization being filed specifically to address nitrosamine risk mitigation issues? If yes, a Level I supplement or a Level III annual notification is expected. A Level III annual notification is accepted only for certain specific changes (refer to number 13). For a Level I supplement, include a summary of root cause investigations and conclusion(s) regarding the confirmed root cause(s) for nitrosamine presence under 3.2.P.2. If no, refer to Post-Notice of Compliance (NOC) changes: Quality Document for the level of the change (Level I or III). Could the proposed changes increase the nitrosamine risk relative to the approved/marketed product? If yesFootnote *, assess the impact of the proposed change on nitrosamine presence and include a risk assessment summary/discussion relative to the approved product in the submission. If noFootnote **, a summary and discussion of a nitrosamine risk assessment is not expected in the submission but do include a brief justification for no increased nitrosamine risk relative to the approved drug product in the notes to reviewers, 3.2.P.2 or 3.2.P.5.
- Appendix 2 footnote *
-
For example, changes to drug substance or drug product manufacturing processes, changes to the drug product composition (API, excipients), changes in dosage form, and changes to the container closure system may increase risk relative to the approved drug product.
- Appendix 2 footnote **
-
For example, the addition of a proportionally-formulated strength would not be expected to increase risk relative to the approved drug product.
Appendix 3: Enhanced Ames assay test conditions
The Organisation for Economic Co-operation and Development (OECD)'s Test Guideline No. 471 "Bacterial Reverse Mutation Test" provides standard recommendations for the conduct of the bacterial reverse mutation test (also known as the Ames assay) to assess the mutagenic potential of a test compound. For N-nitrosamines, enhanced testing conditions for the Ames assay are recommended due to the reported reduced sensitivity of the assay under standard conditions for some N-nitrosamines such as N-nitroso-dimethylamine (NDMA). Moreover, very little is known about the sensitivity of the Ames assay to N-nitrosamine drug substance related impurities (NDSRIs), which are a recently recognized class of N-nitrosamine impurities structurally related to the drug substance. NDSRIs generally have a wider variety of functional groups present than typically found in low molecular weight N-nitrosamines (such as NDMA) historically studied.
If a standard Ames assay is conducted and produces a positive result, there is no need to conduct an additional assay using enhanced testing conditions.
The enhanced Ames assay test conditions presented below are informed by work conducted by FDA's National Center for Toxicological Research (NCTR) (Li et. al., 2023), as well as other groups, and have been evaluated for a variety of N-nitrosamines including NDSRIs. Evaluation of Ames assay test conditions for N-nitrosamines is ongoing with a goal to identify the most robust Ames testing conditions. The enhanced Ames assay test conditions described below will be updated as warranted. Deviations from the recommended conditions should be justified.
Tester strains: S. typhimurium TA98, TA100, TA1535, TA1537, and E. coli WP2 uvrA (pKM101) tester strains should be included.
Type of assay and preincubation time: The pre-incubation, and not plate incorporation, method should be used. The recommended pre-incubation time is 30 minutes.
Species and concentration of S9: Ames assays should be conducted in the absence of a post-mitochondrial fraction (S9), and also in the presence of 30% rat liver S9, as well as 30% hamster liver S9. The rat and hamster post-mitochondrial fractions (S9s) should be prepared from rodents treated with inducers of cytochrome P450 enzymes (for example, a combination of phenobarbital and β-naphthoflavone).
Negative (solvent/vehicle) control: Solvents need to be compatible with the Ames assay as per the OECD 471 guideline. Solvents can include, but are not limited to:
- water
- organic solvents such as acetone, methanol and DMSO
When an organic solvent is used, the lowest possible volume should be included in the pre-incubation mixture with justification to indicate that the volume of solvent does not interfere with metabolic activation of the N-nitrosamine.
Positive controls: Concurrent strain-specific positive controls should be included per the OECD 471 guideline.
Two N-nitrosamines that are known to be mutagenic in the presence of S9 should also be included as positive controls.
The choice of the N-nitrosamine positive controls needs to be justified based on the anticipated metabolism of the N-nitrosamine and the cytochrome P450 enzymes most likely involved. In addition, if an organic solvent is used to dissolve the test compound, it is recommended that the volume of organic solvent employed to dissolve the N-nitrosamine positive controls results in a similar concentration as for the test compound in the pre-incubation mix, if possible.
N-Nitrosamine positive controls to consider include:
- NDMA (CAS # 62-75-9)
- 1-Cyclopentyl-4-nitrosopiperazine (CAS # 61379-66-6)
- An NDSRI
All other recommendations for the Ames assay should follow the OECD 471 guideline.
References:
OECD Test Guideline No. 471 "Bacterial Reverse Mutation Test". 2020
Li et al. Revisiting the mutagenicity and genotoxicity of N-nitroso propranolol in bacterial and human in vitro assays. Regulatory Pharmacology and Toxicology. 2023
Appendix 4: Carcinogenic Potency Categorization Approach (CPCA) for N-nitrosamines
This document describes an approach for assigning an N-nitrosamine impurity (including nitrosamine drug substance-related impurities [NDSRIs]) to a predicted carcinogenic potency category, with a corresponding Acceptable Intake (AI) limit, based on an assessment of activating or deactivating structural features present in the molecule. In the context of this document, activating or deactivating features are defined as molecular substructures that are associated with an increase or decrease, respectively, in carcinogenic potency.
The Carcinogenic Potency Categorization Approach is based on structure-activity relationship (SAR) concepts described in recent scientific publications for N-nitrosamine compoundsFootnote 1 and also used a set of approximately 80 N-nitrosamines with either rat TD50 values from the Carcinogenic Potency Database (CPDB) and/or the Lhasa Carcinogenicity Database (LCDB)Footnote 2, relative potency classifications as defined by Rao et al. (1979)Footnote 3, and/or AI limits based on previously-conducted surrogate analysesFootnote 4. The approach assumes that the α-hydroxylation mechanism of metabolic activationFootnote 5 is responsible for the mutagenic and highly potent carcinogenic response observed for many N-nitrosamines. Structural features that directly increase or decrease the favorability of the activation mechanism, or that increase the clearance of the nitrosamine by other biological pathways, are expected to have a corresponding effect on carcinogenic potency. Therefore, a prediction of the mutagenic potential and carcinogenic potency of an N-nitrosamine can be generated based on its structural features.
It is recognized that the science is evolving in the prediction of mutagenic potential and carcinogenic potency based on SAR concepts. Therefore, the predicted Carcinogenic Potency Categorization Approach described in this document is a conservative approach that represents the best available science at this time and is expected to be further refined and expanded as new data become available. This may include refinement of the AI limits associated with predicted carcinogenic potency categories and changes to the structural features and their associated activating and deactivating feature scores.
The Carcinogenic Potency Categorization Approach applies to N-nitrosamines bearing a carbon atom on both sides of the N-nitroso group, and where the carbon is not directly double bonded to a heteroatom (that is, N-nitrosamides, N-nitrosoureas, N-nitrosoguanidines and other related structures are excluded). Additionally, the potency categorization approach does not apply to N-nitrosamines where the N-nitroso group is within an aromatic ring (for example, nitrosated indole). For N-nitrosamines containing two N-nitroso groups, the group with the highest predicted carcinogenic potency (that is, the group with the lowest numerical potency category) defines the AI for the entire moleculeFootnote 6. The α- and β-carbons are defined relative to the N-nitroso group, as illustrated in Figure 1.
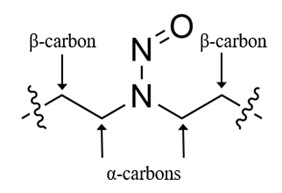
Figure 1 - Text description
Figure 1 shows a partial chemical structure which consists of a nitrogen-nitrogen single bond with an oxygen atom double-bonded to one of the nitrogen atoms. The other nitrogen atom is bonded to two α-carbon atoms, which are each bonded to a β-carbon. Wavy lines beyond each β-carbon atom indicates that the rest of the structure is undefined.
The process for predicting the appropriate carcinogenic potency category is described in Figure 2. Table 1 summarizes the five predicted carcinogenic potency categories and their associated AI limits. Supporting tables to calculate the Potency Score referenced in Figure 2 are in Appendix A and example calculations are presented in Appendix B.
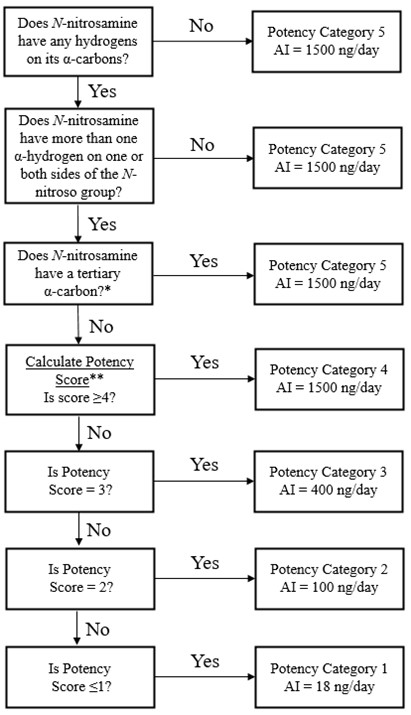
Figure 2 - Text description
Figure 2 shows a flowchart with a step-by-step process of how to assign a potency category to an N-nitrosamine. Each step has two options to choose from, and the steps are followed until a potency category of 1, 2, 3, 4, or 5 is reached. The potency category determines the acceptable intake (AI) limit that the N-nitrosamine impurity can be controlled at.
Does the N-nitrosamine have any hydrogens on it's α-carbons? If no, it is potency category 5 with an AI limit of 1500 ng/day. If yes, does the N-nitrosamine have more than one α-hydrogen on one or both sides of the N-nitroso group? If no, it is potency category 5 with an AI limit of 1500 ng/day. If yes, does the N-nitrosamine have a tertiary α-carbon? A tertiary α-carbon is defined as an α-carbon atom in an sp3 hybridization state, bonded to three other carbon atoms. If yes, it is potency category 5 with an AI limit of 1500 ng/day. If no, calculate the potency score. To calculate the potency score, see Appendix A. Is the potency score ≥4? If yes, it is potency category 4 with an AI limit of 1500 ng/ day. If the potency score is not ≥4, is the potency score=3? If yes, it is potency category 3 with an AI limit of 400 ng/day. If no, is the potency score=2? If yes, it is potency category 2 with an AI limit of 100 ng/day. If no, is the potency score ≤1? If yes, it is potency category 1 with an AI limit of 18 ng/day.
*A tertiary α-carbon is defined as an α-carbon atom in an sp3 hybridization state, bonded to three other carbon atoms.
** To calculate Potency Score, see Appendix A.
| Potency Category | Recommended AI Limit |
Comments |
|---|---|---|
| 1 | 18 | The recommended AI limit of 18 ng/day is equal to the class-specific TTC for N-nitrosamine impurities.Footnote * N-nitrosamines assigned to Category 1 are predicted to have high carcinogenic potency; however, the class-specific TTC for N-nitrosamine impurities is considered sufficiently protective to patients. |
| 2 | 100 | The recommended AI limit of 100 ng/day is representative of two potent, robustly tested N-nitrosamines, N-nitrosodimethylamine (NDMA) and 4-(methylnitrosamino)-1-(3-pyridyl)-1-(butanone) (NNK), which have recommended AI limits of 96 ng/day and 100 ng/day, respectively. N-nitrosamines assigned to Category 2 are predicted to have carcinogenic potency no higher than NDMA and NNK. |
| 3 | 400 | Compared to Potency Category 2, N-nitrosamines in this category have lower carcinogenic potency due to, for example, the presence of a weakly deactivating structural feature. The recommended AI limit was set to reflect a 4-fold decrease in carcinogenic potency from Category 2. |
| 4 | 1500 | N-Nitrosamines assigned to Category 4 may be metabolically activated through an α-hydroxylation pathway but are predicted to be of low carcinogenic potency, for example, because the pathway is disfavored due to steric or electronic influences, or because clearance pathways are favored. The recommended AI limit of 1500 ng/day is set at the TTC per ICH M7.Footnote ** |
| 5 | 1500 | N-Nitrosamines assigned to Category 5 are not predicted to be metabolically activated via an α-hydroxylation pathway due to steric hindrance or the absence of α-hydrogens, or are predicted to form unstable species that will not react with DNA. The recommended AI limit of 1500 ng/day is set at the TTC per ICH M7.Footnote ** |
|
||
Appendix A. Calculation of potency score
For N-nitrosamines not assigned to Potency Category 5, the Potency Score is calculated as the sum of the α-Hydrogen Score (Table 2), Deactivating Feature Score (Table 3) and Activating Feature Score (Table 4) based on selected structural features present in the N-nitrosamine. The N-nitrosamine structure is expected to match exactly one of the α-hydrogen definitions in Table 2, but it may contain multiple or no structural features identified in Tables 3 and 4. In cases where one or more features from Tables 3 and 4 are contained in the N-nitrosamine, the Potency Score should be calculated as outlined in the box below. In cases where the N-nitrosamine contains no features from Tables 3 and 4, the Potency Score will be equal to the α-Hydrogen Score.
Potency Score = α-Hydrogen Score + Deactivating Feature Score (sum all scores for features present in the N-nitrosamine) + Activating Feature Score (sum all scores for features present in the N-nitrosamine)
Count of Hydrogen Atoms on Each |
Example | α-Hydrogen Score |
|---|---|---|
| 0,2 |  |
3Footnote * |
| 0,3 |  |
2 |
| 1,2 | 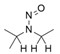 |
3 |
| 1,3 | 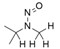 |
3 |
| 2,2 | 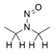 |
1 |
| 2,3 | 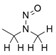 |
1 |
|
||
Table 2 – Text description
The table shows the count of hydrogen atoms on each nitrosamine α-carbon, example structures, and the corresponding α-hydrogen score. For an α-hydrogen count of 0,2 when the methylene α-carbon is not part of an ethyl group, the α-hydrogen score is 3 and when it is part of an ethyl group the score is 2. The example structure includes a nitrogen atom with attached nitroso, phenyl, and propyl groups. For an α-carbon count of 0,3 the α-hydrogen score is 2 and the example structure includes a nitrogen atom with attached nitroso, phenyl, and methyl groups. For an α-hydrogen count of 1,2 the α-hydrogen score is 3 and the example structure includes a nitrogen atom with attached nitroso, isopropyl, and ethyl groups. For an α-hydrogen count of 1,3 the α-hydrogen score is 3 and the example structure includes a nitrogen atom with attached nitroso, isopropyl, and methyl groups. For an α-hydrogen count of 2,2 the α-hydrogen score is 1 and the example structure includes a nitrogen atom with attached nitroso and ethyl groups. For an α-hydrogen count of 2,3 the α-hydrogen score is 1 and the example structure includes a nitrogen atom with attached nitroso, ethyl, and methyl groups.
| Deactivating Feature | Example | Individual Deactivating Feature Score |
|---|---|---|
| Carboxylic acid group anywhere on molecule | 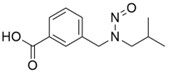 |
+3 |
| N-nitroso group in a pyrrolidine ring |  |
+3 |
| N-nitroso group in a 6-membered ring containing at least one sulfur atom |  |
+3 |
| N-nitroso group in a 5- or 6-membered ringFootnote * |  |
+2 |
| N-nitroso group in a morpholine ring |  |
+1 |
| N-nitroso group in a 7-membered ring | 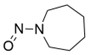 |
+1 |
| Chains of ≥5 consecutive non-hydrogen atoms (cyclic or acyclic) on both side of acyclic N-nitroso group. Not more than 4 atoms in each chain may be in the same ring. |  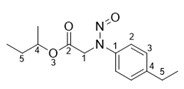 |
+1 |
| Electron-withdrawing groupFootnote ** bonded to α-carbon on only one side of N-nitroso group (cyclic or acyclic) | 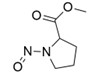 |
+1 |
| Electron-withdrawing groupsFootnote ** bonded to α-carbons on both sides of N-nitroso group (cyclic or acyclic) |  |
+2 |
| Hydroxyl group bonded to β-carbonFootnote *** on only one side of N-nitroso group (cyclic or acyclic) | 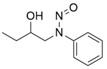 |
+1 |
| Hydroxyl group bonded to β-carbon on both sides of N-nitroso group (cyclic or acyclic) |  |
+2 |
|
||
Table 3 – Text description
The table shows a list of deactivating features, example structures, and associated scores. For the deactivating feature of carboxylic acid group anywhere on the molecule, the deactivating feature score is 3, and the example structure includes a nitrogen atom with an attached nitroso group where one α-carbon atom that is bonded to the nitrogen atom has a 3-carboxyphenyl group attached and the other α-carbon has an isopropyl group attached. For the deactivating feature of N-nitroso group in a pyrrolidine ring, the deactivating feature score is 3, and the example structure includes a pyrrolidine ring with a nitroso group bonded to the nitrogen atom. For the deactivating feature of N-nitroso group in a 6-membered ring containing at least one sulfur atom, the deactivating feature score is 3, and the example structure includes a thiomorpholine ring with a nitroso group bonded to the nitrogen atom. For the deactivating feature of N-nitroso group in a 5- or 6-membered ring, excluding where the N-nitroso group is in a pyrrolidine ring, a 6-membered ring containing at least one sulfur atom, or a morpholine ring, the deactivating feature score is 2, and the example structure includes a piperazine ring with a nitroso group bonded to one of the nitrogen atoms. For the deactivating feature of N-nitroso group in a morpholine ring, the deactivating feature score is 1, and the example structure includes a morpholine ring with a nitroso group bonded to the nitrogen atom. For the deactivating feature of N-nitroso group in a 7-membered ring, the deactivating feature score is 1, and the example structure includes an azepane ring with a nitroso group bonded to the nitrogen atom. For the deactivating feature of chains of ≥5 consecutive cyclic or acyclic non-hydrogen atoms on both sides of an acyclic N-nitroso group where not more than 4 atoms in each chain may be in the same ring, the deactivating feature score is 1, and the example structures include a nitrogen atom with an attached nitroso group where the non-hydrogen atoms beginning with the α-carbons are labelled consecutively with the numbers one through five on both sides of the nitrogen atom. For the deactivating feature of electron-withdrawing group bonded to an α-carbon on only one side of a cyclic or acyclic N-nitroso-group, excluding carboxylic acid and aryl, the deactivating feature score is 1, and the example structure includes a pyrrolidine ring with a nitroso group bonded to the nitrogen atom and a methoxycarbonyl group attached at one α-carbon. For the deactivating feature of electron-withdrawing groups bonded to the α-carbons on both sides of a cyclic or acyclic N-nitroso-group, excluding carboxylic acid and aryl, the deactivating feature score is 2, and the example structure includes a nitrogen atom with an attached nitroso group, a methylsulfonyl group attached to one α-carbon, and an aminocarbonyl group attached to the other α-carbon. For the deactivating feature of hydroxyl group bonded to the β-carbon on only one side of a cyclic or acyclic N-nitroso-group, the β-carbon must be in an sp3 hybridization state and the deactivating feature score is 1, and the example structure includes a nitrogen atom with an attached nitroso group, a 1-hydroxypropyl group attached to one α-carbon, and a phenyl group attached to the other α-carbon. For the deactivating feature of hydroxyl group bonded to both β-carbon on both sides of a cyclic or acyclic N-nitroso-group, the β-carbon must be in an sp3 hybridization state and the deactivating feature score is 2, and the example structure includes a nitrogen atom with an attached nitroso group and 1-hydroxy-2-methylpropyl groups attached to both α-carbon atoms.
Table 4. List of activating features and associated scores. To calculate Activating Feature Score, sum the individual scores for all listed features present in the N-nitrosamine structure. Each activating feature row in the table may only be counted once. Examples are intended to be illustrative only and are not intended to be exhaustive.
| Activating Feature | Example | Individual Activating Feature Score |
|---|---|---|
| Aryl group bonded to α-carbon (i.e., benzylic or pseudo-benzylic substituent on N-nitroso group) |  |
-1 |
| Methyl group bonded to β-carbon (cyclic or acyclic) |  |
-1 |
Table 4 – Text description
The table shows a list of activating features, example structures, and associated scores. For the activating feature of aryl group bonded to α-carbon, the activating feature score is -1, and the example structure includes a nitrogen atom with an attached nitroso group where one α-carbon atom that is bonded to the nitrogen atom has a benzo[b]furan-2-yl group attached and the other α-carbon has an isopropyl group attached. For the activating feature of methyl group bonded to a cyclic or acyclic β-carbon, the activating feature score is -1, and the example structure includes a piperidine ring with a nitroso group attached to the nitrogen atom and a methyl group attached at the 3-position.
Appendix B. Example carcinogenic potency categorization approach calculations based on flow chart
Example 1 shows how the potency categorization approach flow chart (Figure 2) can be applied to the N-nitrosamine, N-nitroso-felodipine. N-Nitroso-felodipine is placed in Potency Category 5 with an associated AI limit of 1500 ng/day.
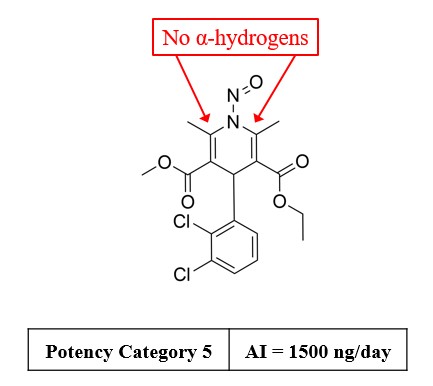
Example 1a - Text description
The image for N-nitroso-felodipine shows the structure includes a dihydropyridine ring with α,β-unsaturation and a nitroso group attached to the nitrogen atom. The dihydropyridine ring is substituted with methyl groups at the 2 and 6 positions, alkoxycarbonyl groups at the 3 and 5 positions, and a 2,3-dichlorophenyl group at the 4 position. Red arrows directed at the two α-carbons indicate that no α-hydrogens are present. The image also shows N-nitroso-felodipine is placed in potency category 5 with an AI limit of 1500 ng/day.
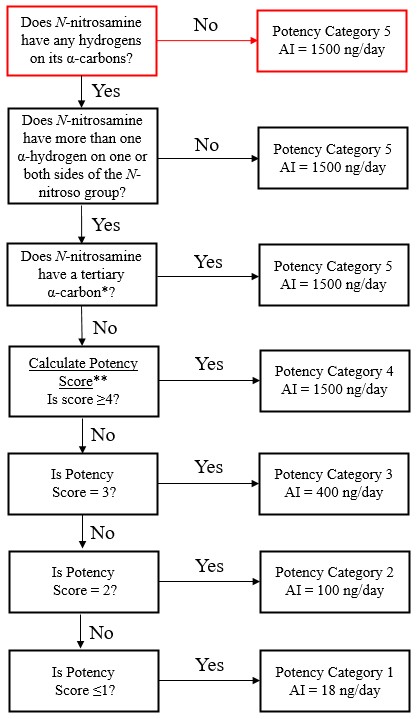
Example 1b - Text description
Example 1b shows Figure 2 with emphasis on the steps for N-nitroso-felodipine: Does the N-nitrosamine have any hydrogens on it's α-carbons? No, therefore it is potency category 5 with an AI=1500 ng/day.
Example 2 shows how the potency categorization approach flow chart (Figure 2) can be applied to the N-nitrosamine, N-nitroso-enalapril. N-Nitroso-enalapril is placed in Potency Category 5 with an associated AI limit of 1500 ng/day.
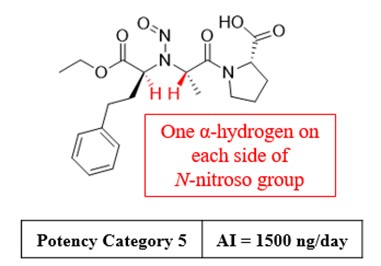
Example 2a - Text description
The image shows the structure for N-nitroso-enalapril includes a nitrogen atom with an attached nitroso group. One α-carbon atom that is bonded to the nitrogen atom has a ethoxycarbonyl group and a 2-phenylethyl group attached. The other α-carbon atom that is bonded to the nitrogen atom has a methyl group and a proline-N-carbonyl group attached. The two α-carbon atoms each have one α-hydrogen atom attached which are highlighted with red colour and text indicating one α-hydrogen on each side of N-nitroso group. The image also shows N-nitroso-enalapril is placed in potency category 5 with an AI limit of 1500 ng/day.
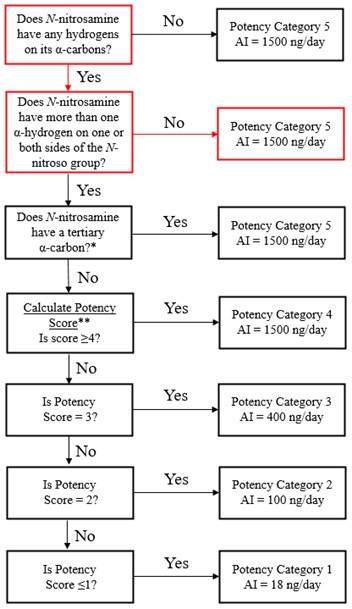
Example 2b - Text description
Example 2b shows Figure 2 with emphasis on the steps for N-nitroso-enalapril: Does the N-nitrosamine have any hydrogens on it's α-carbons? Yes. Does the N-nitrosamine have more than one α-hydrogen on one or both sides of the N-nitroso group? No, therefore it is potency category 5 with an AI limit of 1500 ng/day.
Example 3 shows how the potency categorization approach flow chart (Figure 2) can be applied to the N-nitrosamine, N-nitroso-ketamine. N-Nitroso-ketamine is placed in Potency Category 5 with an associated AI limit of 1500 ng/day.
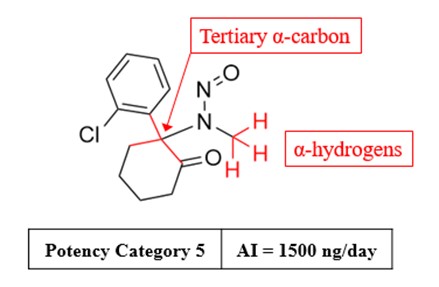
Example 3a - Text description
The image shows the structure for N-nitroso-ketamine includes a nitrogen atom with an attached nitroso group. One α-carbon that is bonded to the nitrogen atom has a methyl group attached with the three α-hydrogen atoms highlighted in red colour, and text indicating that they are α-hydrogens. The other α-carbon that is bonded to the nitrogen atom is part of cyclohexanone ring, has a 2-chlorophenyl group attached to it, and an arrow pointing toward it with text indicating that it is a tertiary α-carbon atom. The image also shows N-nitroso-ketamine is placed in potency category 5 with an AI limit of 1500 ng/day.
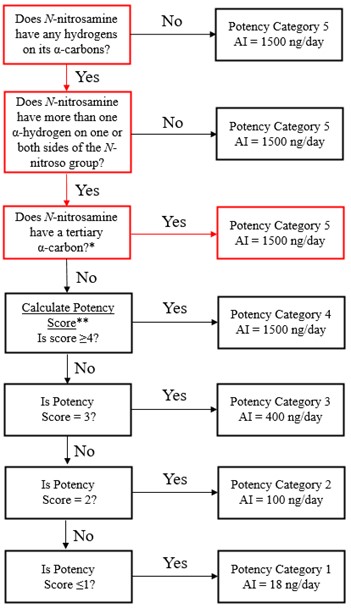
Example 3b - Text description
Example 3b shows Figure 2 with emphasis on the steps for N-nitroso-ketamine: Does the N-nitrosamine have any hydrogens on it's α-carbons? Yes. Does the N-nitrosamine have more than one α-hydrogen on one or both sides of the N-nitroso group? Yes. Does the N-nitrosamine have a tertiary α-carbon? Yes, therefore it is potency category 5 with an AI limit of 1500 ng/day.
Example 4 – N-Nitroso-nebivolol
Example 4 shows how the potency categorization approach flow chart (Figure 2) can be applied to the N-nitrosamine, N-nitroso-nebivolol. A Potency Score of 4 is calculated for N-nitroso-nebivolol, resulting in its placement in Potency Category 4 with an associated AI limit of 1500 ng/day.
Count of α-Hydrogens |
Score |
Feature highlighted in red |
|---|---|---|
| 2,2 | 1 |  |
Deactivating features |
Score |
Feature highlighted in red |
|---|---|---|
| Hydroxyl group bonded to β-carbons on both sides of N-nitroso group (cyclic or acyclic) | +2 |  |
| Chains of ≥5 consecutive non-hydrogen atoms (cyclic or acyclic) on both sides of acyclic N-nitroso group. Not more than 4 atoms in each chain may be in the same ring. | +1 |  |
No Activating Features Present |
Potency score = 1 + 2 + 1 = 4 |
Potency category 4 |
AI = 1500 ng/day |
Example 4a - Text description
The table shows how to calculate a potency score for N-nitroso-nebivolol. The count of α-hydrogens is 2,2 which has a score of 1, and the structure of N-nitroso-nebivolol is highlighting the two hydrogen atoms present on both α-carbon atoms in red colour. For the deactivating feature of hydroxyl group bonded to β-carbons on both sides of the N-nitroso group, the score applied is 2, and the structure of N-nitroso-nebivolol shown is highlighting the two hydroxyl groups present on both β-carbon atoms in red colour. For the deactivating feature of chains of ≥5 consecutive non-hydrogen atoms on both sides of an acyclic N-nitroso group, the score applied is 1, and the structure of N-nitroso-nebivolol shown is highlighting the five non-hydrogen atom bonds that extend outward from the N-nitroso group in red colour. The potency score, 4, is shown as the sum of the α-hydrogen and deactivating feature scores (1+2+1=4). The image also shows N-nitroso-nebivolol is placed in potency category 4 with an AI limit of 1500 ng/day.
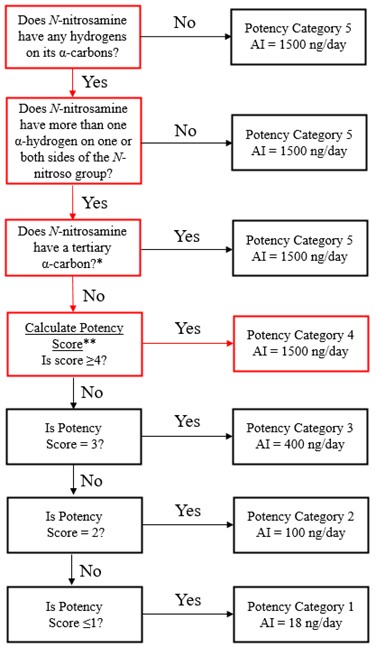
Example 4b - Text description
Example 4b shows Figure 2 with emphasis on the steps for N-nitroso-nebivolol: Does the N-nitrosamine have any hydrogens on it's α-carbons? Yes. Does the N-nitrosamine have more than one α-hydrogen on one or both sides of the N-nitroso group? Yes. Does the N-nitrosamine have a tertiary α-carbon? No. Calculate the potency score. Is the score ≥4? Yes, therefore it is potency category 4 with an AI limit of 1500 ng/day.
Example 5 – N-Nitroso-meropenem
Example 5 shows how the potency categorization approach flow chart (Figure 2) can be applied to the N-nitrosamine, N-nitroso-meropenem. A Potency Score of 4 is calculated for N-nitroso-meropenem, resulting in its placement in Potency Category 4 with an associated AI limit of 1500 ng/day.
Count of α-Hydrogens |
Score |
Feature highlighted in red |
|---|---|---|
| 1,2 | 3 |  |
Deactivating features |
Score |
Feature highlighted in red |
|---|---|---|
| Carboxylic acid group anywhere on molecule | +3 |  |
| N-nitroso group in a pyrrolidine ring | +3 |  |
| Electron-withdrawing group bonded to α-carbon on only one side of N-nitroso group (cyclic or acyclic) | +1 |  |
No Activating Features Present |
Potency score = 3 + 3 + 3 + 1 = 10 |
Potency category 4 |
AI = 1500 ng/day |
Example 5a - Text description
The table shows how to calculate a potency score for N-nitroso-meropenem. The count of α-hydrogens is 1,2 which has a score of 3, and the structure of N-nitroso-nebivolol is highlighting the two hydrogen atoms present on one α-carbon atom and the one hydrogen atom present on the other α-carbon atom in red colour. For the deactivating feature of carboxylic acid group anywhere in the structure, the score applied is 3, the structure of N-nitroso-meropenem shown is highlighting the carboxylic acid group which is present in red colour. For the deactivating feature of N-nitroso group in a pyrrolidine ring, the score applied is 3, and the structure of N-nitroso-meropenem shown is highlighting the five-membered pyrrolidine ring with a N-nitroso group attached in red colour. For the deactivating feature of electron-withdrawing group bonded to α-carbon on only one side of a N-nitroso group, the score applied is 1, and the structure of N-nitroso-meropenem shown is highlighting a dimethylaminocarbonyl group attached to an α-carbon in red colour. The potency score, 4, is shown as the sum of the α-hydrogen and deactivating feature scores (3+3+3+1=10). The image also shows N-nitroso-meropenem is placed in potency category 4 with an AI limit of 1500 ng/day.
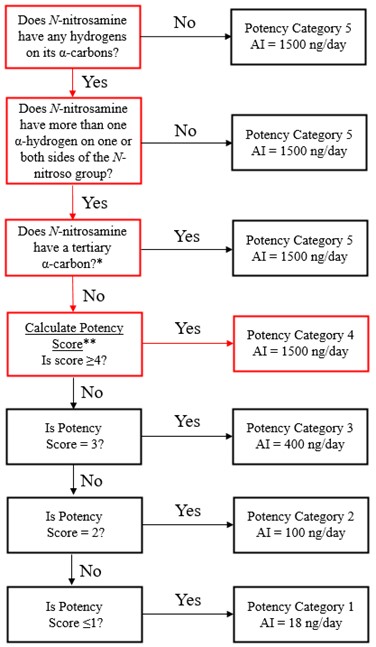
Example 5b - Text description
Example 5b shows Figure 2 with emphasis on the steps for N-nitroso-meropenem: Does the N-nitrosamine have any hydrogens on it's α-carbons? Yes. Does the N-nitrosamine have more than one α-hydrogen on one or both sides of the N-nitroso group? Yes. Does the N-nitrosamine have a tertiary α-carbon? No. Calculate the potency score. Is the potency score ≥4? Yes, therefore it is potency category 4 with an AI limit of 1500 ng/day.
Example 6 – N-Nitroso-desloratadine
Example 6 shows how the potency categorization approach flow chart (Figure 2) can be applied to the N-nitrosamine, N-nitroso-desloratadine. A Potency Score of 3 is calculated for N-nitroso-desloratadine, resulting in its placement in Potency Category 3 with an associated AI limit of 400 ng/day.
Count of α-Hydrogens |
Score |
Feature highlighted in red |
|---|---|---|
| 2,2 | 1 |  |
Deactivating features |
Score |
Feature highlighted in red |
|---|---|---|
| N-nitroso group in a 5- or 6-membered ring | +2 |  |
No Activating Features Present |
| Potency score = 1 + 2 = 3 | Potency category 3 | AI = 400 ng/day |
Example 6a - Text description
The table shows how to calculate a potency score for N-nitroso-desloratadine. The count of α-hydrogens is 2,2 which has a score of 1, and the structure of N-nitroso-desloratadine is highlighting the two hydrogen atoms present on both α-carbon atoms in red colour. For the deactivating feature of N-nitroso group in a 5- or 6-membered ring, the score applied is 2, and the structure of N-nitroso-loratadine shown is highlighting the six-membered piperidine ring with a N-nitroso group attached in red colour. The potency score 3, is shown as the sum of the α-hydrogen and deactivating feature scores (1+2=3). The image also shows N-nitroso-desloratadine is placed in potency category 3 with an AI limit of 400 ng/day.
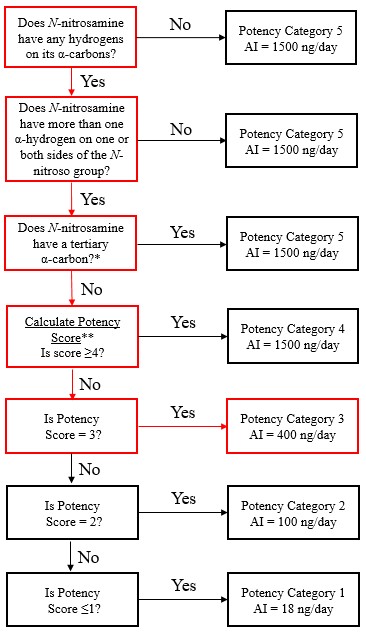
Example 6b - Text description
Example 6b shows Figure 2 with emphasis on steps for N-nitroso-desloratadine: Does the N-nitrosamine have any hydrogens on it's α-carbons? Yes. Does the N-nitrosamine have more than one α-hydrogen on one or both sides of the N-nitroso group? Yes. Does the N-nitrosamine have a tertiary α-carbon? No. Calculate the potency score. Is the potency score ≥4? No. Is the potency score = 3? Yes, therefore it is potency category 3 with an AI limit of 400 ng/day.
Example 7 – N-Nitroso-sertraline
Example 7 shows how the potency categorization approach flow chart (Figure 2) can be applied to the N-nitrosamine, N-nitroso-sertraline. A Potency Score of 2 is calculated for N-nitroso- sertraline, resulting in its placement in Potency Category 2 with an associated AI limit of 100 ng/day.
Count of α-Hydrogens |
Score |
Feature highlighted in red |
|---|---|---|
| 1,3 | 3 |  |
No Deactivating Features Present |
Activating features |
Score |
Feature highlighted in red |
|---|---|---|
| Aryl group bonded to α-carbon (i.e., benzylic or pseudo-benzylic substituent on N-nitroso group) | -1 |  |
Potency score = 3 - 1 = 2 |
Potency category 2 |
AI = 100 ng/day |
Example 7a - Text description
The table shows how to calculate a potency score for N-nitroso-sertraline. The count of α-hydrogens is 1,3 which has a score of 3, and the structure of N-nitroso-sertraline is highlighting the single hydrogen atom present on one α-carbon atom and the 3 hydrogen atoms on the other α-carbon atom in red colour. For the activating feature of aryl group bonded to α-carbon, the score applied is -1 and the structure of N-nitroso-sertraline shown is highlighting the aryl group attached to one α-carbon in red colour. The potency score 2, is shown as the sum of the α-hydrogen and deactivating feature scores (3-1=2). The image also shows N-nitroso-sertraline is placed in potency category 2 with an AI limit of 100 ng/day.
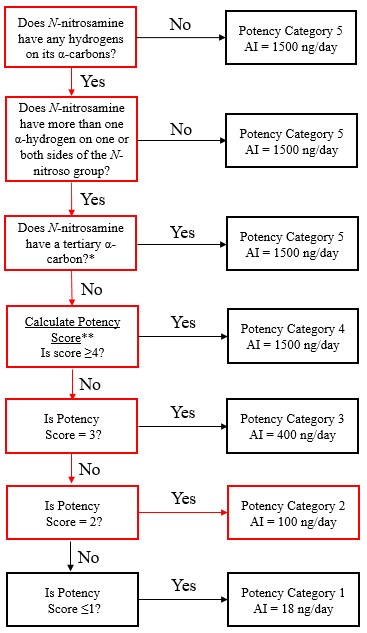
Example 7b - Text description
Example 7b shows Figure 2 with emphasis on steps for N-nitroso-sertraline: Does the N-nitrosamine have any hydrogens on it's α-carbons? Yes. Does the N-nitrosamine have more than one α-hydrogen on one or both sides of the N-nitroso group? Yes. Does the N-nitrosamine have a tertiary α-carbon? No. Calculate the potency score. Is the potency score ≥4? No. Is the potency score = 3? No. Is the potency score = 2? Yes, therefore it is potency category 2 with an AI limit of 100 ng/day.
Example 8 – N-Nitroso-lorcaserin
Example 8 shows how the potency categorization approach flow chart (Figure 2) can be applied to the N-nitrosamine, N-nitroso-lorcaserin. A Potency Score of 1 is calculated for N-nitroso-lorcaserin, resulting in its placement in Potency Category 1 with an associated AI limit of 18 ng/day.
Count of α-Hydrogens |
Score |
Feature highlighted in red |
|---|---|---|
| 2,2 | 1 |  |
Deactivating features |
Score |
Feature highlighted in red |
|---|---|---|
| N-nitroso group in a 7-membered ring | +1 |  |
Activating Features |
Score |
Feature highlighted in red |
|---|---|---|
| Methyl group bonded to β-carbon (cyclic or acyclic) | -1 |  |
Potency score =1 + 1 - 1 = 1 |
Potency category 1 |
AI = 18 ng/day |
Example 8a - Text description
The table shows how to calculate a potency score for N-nitroso-lorcaserin. The count of α-hydrogens is 2,2 which has a score of 1, and the structure of N-nitroso-lorcaserin is highlighting the 2 hydrogen atoms present on both α-carbon atoms in red colour. For the deactivating feature of N-nitroso group in a 7-membered ring, the score applied is 1 and the structure of N-nitroso-lorcaserin shown is highlighting bonds comprising the seven-membered ring with a nitroso group attached in red colour. For the activating feature of methyl group bonded to β-carbon, the score applied is -1 and the structure of N-nitroso-lorcaserin shown is highlighting the methyl group attached to one β-carbon in red colour. The potency score 1 is shown as the sum of the α-hydrogen, deactivating, and activating feature scores (1-1+1=1). The image also shows N-nitroso-lorcaserin is placed in potency category 1 with an AI limit of 18 ng/day.
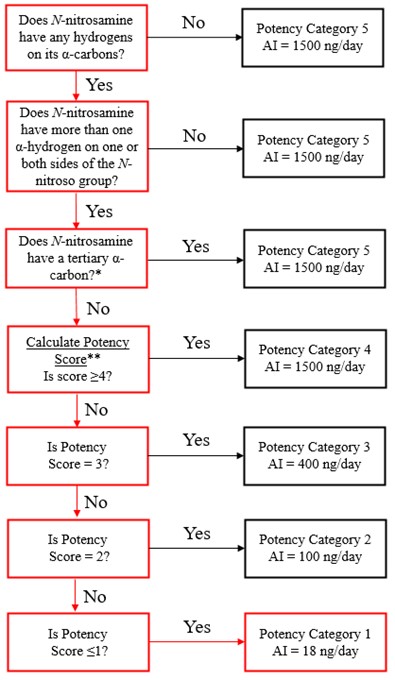
Example 8b - Text description
Example 8b shows Figure 2 with emphasis on steps for N-nitroso-lorcaserin: Does the N-nitrosamine have any hydrogens on it's α-carbons? Yes. Does the N-nitrosamine have more than one α-hydrogen on one or both sides of the N-nitroso group? Yes. Does the N-nitrosamine have a tertiary α-carbon? No. Calculate the potency score. Is the potency score ≥4? No. Is the potency score = 3? No. Is the potency score = 2? No. Is the potency score ≤ 1? Yes, therefore it is potency category 1 with an AI limit of 18 ng/day.
- Footnote 1
-
For example, see Cross KP and Ponting DJ, 2021. Developing Structure-Activity Relationships for N-Nitrosamine Activity, Comput Toxicol, 20:100186; Thomas R, Tennant RE, Oliveira AAF, and Ponting DJ, 2022. What Makes a Potent Nitrosamine? Statistical Validation of Expert-Derived Structure-Activity Relationships, Chem Res Toxicol, 35:1997–2013; and Ponting DJ, Dobo KL, Kenyon MO, and Kalgutkar AS, 2022. Strategies for Assessing Acceptable Intakes for Novel N-Nitrosamines Derived From Active Pharmaceutical Ingredients, J Med Chem, 65:15584–15607.
- Footnote 2
-
See Lhasa Carcinogenicity Database at https://carcdb.lhasalimited.org/.
- Footnote 3
-
Rao TK, Young JA, Lijinsky W and Epler JL, 1979. Mutagenicity of Aliphatic Nitrosamines in Salmonella typhimurium, Mutat Res, 66:1-7.
- Footnote 4
-
Questions and answers for marketing authorisation holders / applicants on the CHMP opinion for the Article 5(3) referral.
- Footnote 5
-
Li Y, Hecht SS, 2022. Metabolic Activation and DNA Interactions of Carcinogenic N-Nitrosamines to Which Humans Are Commonly Exposed, Int J Mol Sci, 23:4559.
- Footnote 6
-
For N-nitrosamines containing more than two N-nitroso groups, the applicant or manufacturer should contact the applicable drug regulatory authority for further guidance.
Appendix 5: Bioequivalence studies for reformulated drug products (new)
In general, qualitative changes to a drug product should be supported by a demonstration of bioequivalence of the reformulated product in comparison to the unmodified formulation, or to the Canadian reference product (CRP) for subsequent-entry (generic) drug products, in an in vivo comparative bioavailability study.
Reformulated immediate-release, solid oral or oral suspension dosage forms that contain Biopharmaceutics Classification System (BCS) Class I (high solubility, high permeability) drug substances are eligible for a waiver of the requirement to provide a bioequivalence study (biowaiver), per the recommendations detailed in the ICH M9 harmonised guideline Biopharmaceutics Classification System-based biowaivers. Additionally, a biowaiver may be considered for drug products with BCS Class III (high solubility, low permeability) drug substances that are reformulated to include specific antioxidants (i.e., ascorbic acid, α-tocopherol, propyl gallate, or cysteine hydrochloride). Recent studies have demonstrated that the incorporation of small quantities of these antioxidants in BCS III model drugs do not alter the permeability of the drug substance or the activity of intestinal transporters.Footnote 1 Footnote 2 Consequently, the addition of these specific antioxidants is not expected to impact the absorption or bioavailability of the drug. The results of these studies and conclusions can be extrapolated to drug substances that exhibit high permeability, given the minimal effect on the BCS Class III (low permeability) model drug substances. Therefore, a biowaiver may also be considered for drug products with BCS Class II (low solubility, high permeability) drug substances that are reformulated to include these specific antioxidants (i.e., ascorbic acid, α-tocopherol, propyl gallate, or cysteine hydrochloride).
As previously noted, a pH modifier (for example, sodium carbonate) that alters the gastrointestinal microenvironment to neutral or basic pH, may be employed to inhibit nitrosamine impurity formation. The addition of a pH modifier would not be expected to impact the permeability of a drug substance with amine functional groups. As such, biowaiver requests may be considered for drug products that contain BCS Class I, II, or III drug substances, that are reformulated to add a pH modifier.
A biowaiver for a reformulated drug product should be supported by a scientific justification of the choice and amount of the antioxidant or pH modifier employed in the formulation. In addition, the manufacturing process should remain generally unchanged apart from the addition of the new excipient.
The ICH M9 guideline provides recommendations to support the biopharmaceutics classification of drug substances and details regarding the criteria and data requirements for a biowaiver based on BCS principles.
Biowaiver requests for reformulated immediate-release, solid oral or oral suspension dosage forms that contain BCS Class IV (low solubility, low permeability) drug substances and reformulated modified-release products should be supported by robust approaches, such as a validated in vitro-in vivo correlation (IVIVC) or physiologically based biopharmaceutics modeling (PBBM). Biowaivers may be considered on a case-by-case basis for reformulated enteric-coated drug products that contain BCS Class I, II, or III drug substances when scientifically justified. If a biowaiver is not applicable, comparative bioavailability studies should be provided to support the change in formulation.
The Health Canada guidance documents, Conduct and Analysis of Comparative Bioavailability Studies and Comparative Bioavailability Standards: Formulations Used for Systemic Effects, should be consulted for study design and statistical analysis considerations, and for recommendations on the standards that should be met to determine bioequivalence, respectively.
- Footnote 1
-
Kulkarni CP, Yang J, Koleske ML, Lara G, Alam K, Raw A, Rege B, Zhao L, Lu D, Zhang L, Yu LX, Lionberger RA, Giacomini KM, Kroetz DL, and Yee SW, 2024. Effect of Antioxidants in Medicinal Products on Intestinal Drug Transporters, Pharmaceutics, 16:647-659. https://doi.org/10.3390/pharmaceutics16050647.
- Footnote 2
-
Lu, D, Rege B, Raw A, Yang J, Alam K, Bode C, Zhao L, Faustino P, Wu F, Shakleya D, Nickum E, Li BV, Wang R, Stier E, Miezeiewski B, Patel R, Boam A, Lionberger R, Keire D, and Yu L, 2024, Antioxidants Had No Effects on the In-Vitro Permeability of BCS III Model Drug Substances, J Pharm Sci, 113: 2708-2714. https://doi.org/10.1016/j.xphs.2024.05.033.