
Draft guidance on how to interpret ‘significant change’ of a medical device: Types of changes
On this page
- Changes to manufacturing processes, facilities or equipment
- Changes to manufacturing quality control procedures
- Changes in design
- Changes to sterilization
- Changes to software
- Changes in materials for non in vitro diagnostic devices
- Changes in materials for in vitro diagnostic devices
- Changes to labelling
- Class III and IV amendments for compatible Class II devices
- Changes to diagnostic ultrasound systems
Changes to manufacturing processes, facilities or equipment
For the purposes of this section, a manufacturing change impacts how a device is made, but does not intentionally change the device or its packaging and labelling material.
Examples of a manufacturing change would be:
- changes to the component formation process
- for example, from an extruded to a machined component
- altering manufacturing machine tolerances
- replacing equipment used to perform a manufacturing function
- moving manufacturing to a new facility
A change to the manufacturing process, facility or equipment can impact the safety or effectiveness of a device. You should consider the impact of all manufacturing changes on the device specifications, performance and material properties. Evaluate these changes using the other applicable sections of this guidance document.
If a change to the manufacturing process, facility or equipment involves changes to the sterilization process, refer to the section on changes to sterilization.
Tolerances that have been increased to allow for more variation in the end product is likely a significant change, as it may impact device performance.
Changes that are likely non-significant are as follows:
- increases to the tolerance ranges if the impact of the change can be interpolated from the verification or validation testing previously submitted and reviewed by Health Canada for the authorized device
- Submitted information must demonstrate the safety and effectiveness of the device over the full range of allowable outputs from a new, wider tolerance range.
- changes that tighten specifications within the existing range of authorized specifications where the mean value is not changed
- changes to the manufacturing process, facility or equipment that do not impact the device specifications, performance or materials and for which incoming inspections to evaluate the material provided by the supplier have not been changed
- the addition of a new manufacturing facility when:
- the manufacturer's name and address on the device labelling stays the same and
- the new facility has the same manufacturing process and equipment of the same specification
- the new facility is not a new sterilization facility or new abattoir for animal-derived materials
For more information, refer to the following sections:
- Changes to sterilization
- Changes in materials for non in vitro diagnostic devices
For combination products, there are increased sensitivities associated with the production and application of active pharmaceutical ingredients (APIs). As changes to the manufacturing process of a combination product that involve APIs are often significant, we recommend that you contact the Medical Devices Directorate by email at meddevices-instrumentsmed@hc-sc.gc.ca for assistance.
Table of examples
| Device | Proposed change | Significant or not significant |
|---|---|---|
Devices sold as non-sterile |
Packaging changed from 1 variant of polyethylene to another due to supplier rationalization or cost-saving measures. Verification and stability testing shows integrity has not been compromised. |
Not significant |
Drug eluting stent |
A change to a manufacturing site where a polymer or drug coating is applied to the device. |
Significant |
Catheters |
Change in supplier that extrudes the polymer tubing with no change in finished product performance specifications. |
Not significant |
Implantable 3D printed devices |
Changes to additive manufacturing processes including equipment, raw materials and post-printing processes of the device. For details, refer to the guidance document Supporting evidence for implantable medical devices manufactured by 3D Printing. |
Significant |
All devices |
A change in material supplier but the design and/or performance specifications of the finished device are not impacted. |
Not significant |
Biological devices |
Change in animal or human tissue supplier. Note: A change in supplier could impact biological safety requirements for animal or human-derived tissue materials. For more guidance, refer to the section Changes in materials for non in vitro diagnostic devices. |
Significant |
Changes to manufacturing quality control procedures
Changing or adding new testing or inspection activities for incoming materials, in-process materials and final products is not a significant change if the specifications or acceptance criteria have not been loosened and acceptance quality limits (AQL) have not increased. On the other hand, removing or modifying these activities where the result is that specifications or criteria are loosened or the AQLs are increased is a significant change.
Changes to quality control inspections or tests used to control the quality, purity or sterility of materials and devices that affect the specifications of the finished device or its components, including its packaging, are often considered significant. However, if adding or modifying the inspection or test method provides equivalent or improved assurance of reliability, then the modification is not significant. It must be reported at annual renewal, with applicable justification made to support this modification.
Examples include changes to the:
- test or inspection criteria
- test method
- parameter tested or inspected
- statistical rationale or sample size
- acceptance quality limits (AQL)
- validation endpoints
- sterility assurance level (SAL)
For example, changes to the manufacturer's requirements for material acceptance criteria can be considered a significant change if these changes alter the design specifications of the device.
Table of examples
| Device | Proposed change | Significant or not significant |
|---|---|---|
All devices |
Modification or removal of a test that characterizes the final product or its performance (for example, bending test for a metal component) |
Significant |
All devices |
Change in design specifications of a device to accommodate a loosening of acceptance criteria as a result of a change in supplied parts or materials. |
Significant |
All devices |
In-process inspection of tubing length in a catheter removed and a final 100% inspection of tubing length introduced later in the manufacturing process. |
Not significant |
Changes in design
Changes in design range from minor design specification or engineering changes to major changes in operating principles. All design changes must be evaluated, verified and validated according to the accepted procedures recorded in the quality management system.
Significant design changes can include modifications to the:
- device’s control mechanism
- device’s operating principles
- device’s design specifications
- device’s components or accessories
- human factors of the patient or user interface
Control mechanism
A control mechanism is the means by which the action of a device is directed or the output of a device is regulated. As control mechanisms can be complex, any changes to them could alter how the device works. This can impact its risk profile, as well as its safety and effectiveness and therefore changes to the control mechanisms are significant.
Examples of changes:
- pneumatic to electronic control of a mechanical ventilator
- analog to digital control of a device
Operating principles
Operating principles are the means by which a device produces or brings about an intended or appropriate effect. Changes to operating principles are considered significant.
Examples of operating principle changes include switching:
- an energy input source from AC to DC or battery
- an output source from microwave to radiofrequency
- from wired to wireless communication (in relation to therapeutic or diagnostic features)
- between types of wireless communication, such as WIFI to Bluetooth
Examples of operating principle changes for an in vitro diagnostic device (IVDD) include:
- a change from an immunofluorescence-based assay to an enzyme-linked immunosorbent assay (ELISA)
- changes in methods such as specimen pretreatment, incubation times and temperatures, which may affect the operating principle of an IVDD
- are considered significant changes if they result in altered performance characteristics that are reflected in the labelling
For more information on operating principle changes that include software, refer to the section on changes to software.
Design specifications
Changes to design specifications can involve:
- a device’s physical characteristics such as the shape, size, dimension or materials, including non IVDDs and IVDDs
- performance or technical specifications
- patient or user interfaces
- software or firmware updates
For an IVDD, design specifications also include performance characteristics (for example, precision, accuracy, sensitivity, specificity, stability).
A change to design specifications may be significant if it influences 1 of the following 3 factors:
- the indications for use/intended use
- the requirements to have clinical data
- the risk profile of the device
1) Indications for use/intended use
Changes to design that affect the indications for use or intended use are significant. They can impact the target patient population (for example, elderly or pediatric patients), the target anatomical areas or the target disease states.
Example of a significant change:
- changing the length or diameter of a cardiovascular stent so it can be placed in different arteries may introduce an additional indication for use (whether explicitly claimed in the labelling or not)
2) Clinical data
Changes to the design specifications that require new clinical data to validate the safety and/or effectiveness of the device are significant.
Examples of a significant change:
- making a device smaller so that a pediatric indication can be added requires clinical evidence
- introducing a change that results in a new indication or clinical use or introducing a new feature whose use is not well established for that type of device requires new clinical validation
3) Risk profile
You should conduct a risk assessment when making a design change. The risk assessment will identify any new risks. If the risk assessment finds that the change would alter the safety and effectiveness of the device, either positively or negatively, the change is significant.
Examples of a significant change:
- the design change may:
- introduce a new hazard or hazardous situation
- change the severity or the likelihood of the harm occurring
- introduce new required risk mitigation actions
The change is likely not significant if:
- the risk assessment conclusions are not altered by the design change and
- the device performs the same as the original design submitted in a previous application
Components or accessories, including Class II
Adding or changing device components or accessories used with the authorized device is a significant change if it:
- affects the safety, function or compatibility profile of the device as a whole or
- could cause the device to be used in a new way
Modifying an existing or adding a new component or accessory that is intended to be, or labelled to be, compatible with a class III or IV device is a significant change if:
- this addition or modification may impact the safety or effectiveness of the assembled combination of devices, components or accessories
This is true regardless of the classification of the component or accessory, or whether all parts are on the same authorization or authorized separately.
An application to amend the associated Class III or IV device(s) would be required. You would need to:
- describe and validate the change
- verify the safety, effectiveness, compatibility and risk profile of the assembled combination of devices, components and accessories is the same
For more information, refer to the section on Class III and IV amendments for compatible Class II devices.
Human factors of the patient or user interface
A design change that affects how the device is used or how it interacts or interfaces with the patient or user is likely significant.
Examples are changes to:
- the display or layout of a control panel or graphical user interface (GUI)
- for example, a blood glucose meter
- the way the device interacts with the patient or user
- for example, the way a CPAP mask attaches to a patient’s face
- the way a surgical tool or delivery system is designed to be held or used by the surgeon
Cosmetic changes that:
- do not affect function or clinical use are likely not significant
- alter how a device functions or performs clinically are significant (could affect its safety and effectiveness)
Table of examples
| Device | Proposed change | Significant or not significant |
|---|---|---|
All devices |
New foot switch (where there was not one before) added to an electrosurgical generator or other device, and corresponding software added to the operating console. |
Significant |
Non-active surgically invasive devices |
Change in the design characteristics allows for additional or broader indications for use (for example, a smaller-sized hip prosthesis or fracture fixation screw that differs significantly from the previous designs). |
Significant |
Catheters |
Change to the cable design and grip of a steerable ablation catheter, which results in improved deliverability and improved procedural times. |
Significant |
Catheters |
Change to the grip of a steerable ablation catheter to improve comfort for the healthcare professional or to improve the appearance of the device without changing its functionality or any critical forces that could be applied and felt (tactile) by the user. |
Not significant |
Endocardial lead |
Additional polymer support clip added to prevent the electrical connection from becoming dislodged and to increase the axial retention forces. |
Significant |
Ultrasound transducer |
Design of the grip portion updated to improve user comfort. Change does not affect safety or performance. |
Not significant |
Hemofiltration system, including software controls |
New component, combined filter and disposable cartridge added. |
Significant |
Transurethral thermal system for treating benign prostatic hyperplasia |
Software changed to provide automatic control of ramping power, respond to elevated rectal temperatures automatically and adjust power. |
Significant |
Metallic biliary stent for treating malignant strictures |
Two new stent lengths added that are outside the range of previously authorized stent lengths on the same authorization. |
Significant |
Metallic biliary stent for treating malignant strictures |
Two new stent lengths added that are between the previously authorized stent lengths on the same authorization, with no other differences. |
Not significant |
Total knee system |
Longer femoral augments added. |
Significant |
Total hip system |
New bearing surface added. |
Significant |
Acetabular cups |
Change in design to give additional flexibility to implanting surgeons. More holes added to the cups. |
Significant |
Bone void fillers and putty |
Amount of cancellous bone material in the filler increased. |
Significant |
Anaesthesia machine |
Change in the sensor controlling the fresh air proportions. |
Significant |
Automatic implanted cardiac defibrillator |
Internal components, including the capacitors, telemetry coils, batteries and transformers, altered to improve how the device operates. |
Significant |
Cardiac pacing leads |
Two or more electrodes, or a new anchoring mechanism that can result in new indications for use, added and performance claims enhanced. |
Significant |
Pacing lead |
Size of the wire diameter reduced to reduce the overall lead diameter to facilitate introduction into the vessel. |
Significant |
Patent foramen ovale (PFO) closure device |
An 18-millimetre (mm) PFO closure device added to an authorization that includes a 16-mm and a 20-mm PFO closure device. Basic design and delivery system are the same. |
Not significant |
IVDD test kit |
Sample matrix for an IVDD test kit changed from a venous blood sample to a dried blood spot. |
Significant |
Clinical chemical analyzer |
Throughput changed. |
Significant |
Clinical chemical analyzer |
Test volume changed. |
Significant |
Clinical chemical analyzer |
Full automation changed. |
Significant |
Blood glucose monitor |
A new control added. |
Significant |
Blood glucose monitor |
Sample volume reduced by changing the electrode layout, which reduces the test strip sample chamber volume. |
Significant |
Blood glucose monitor |
Alternate sampling location added (for example, abdomen). |
Significant |
Automated ELISA analyzer |
New analyte added (for example, Hepatitis B surface antigen). |
Significant |
IVDD test kit |
The blood collection method for a point-of-care IVDD changes from a capillary draw to a mechanical draw pipette. |
Significant |
Changes to sterilization
Changes that could affect the effectiveness of the sterilization process or the safety of a sterile device are significant.
The nature of sterilization is such that it is impossible to determine by inspection and testing if the device has been sterilized successfully. Medical devices are considered sterile if you can demonstrate a sterility assurance level (SAL) of 10-6 or better. The sterilization process must be verified and validated, and its performance routinely monitored.
Examples of significant sterilization changes are those where the:
- manufacturing process, environment or device material introduces an organism that is more difficult to kill compared to the challenge organism previously used in sterilization validation testing
- manufacturing process, environment or device material increases the bioburden above the maximum bioburden level previously validated in sterilization validation testing
- device design introduces a feature or alters existing features that result in a new worst case most-difficult-to-sterilize location for sterilization validation
- device material is more difficult to sterilize or affects sterilant residuals
- quality control verification and validation processes are changed, such as introducing or removing parametric release or changing the sterilization dose auditing method
- sterilization method is changed
- critical cycle parameters (such as ethylene oxide gas concentration or radiation dose) are changed
- dose delivery is changed (such as changes in loading density or configuration)
In general, a change that triggers a new or increased risk or produces unexpected results from routine verification and validation is a significant change. Adding a new test acceptance criteria or test method to the existing process to provide the same or better assurance of sterility, reliability or similar safety aspects is a non-significant change.
Non-significant changes would be those limited to:
- adding a new facility, chamber or equipment that uses the same sterilization method and critical cycle parameters (for example, EO gas concentration or radiation dose) and validating to a SAL of 10-6 using a Health Canada-recognized standard (for example, in ISO 11135 or ISO 11137)
Examples:
- a contract sterilizer with multiple locations moves sterilization to a different facility using a verified equivalent sterilization process
- equipment changes (within an approved site or at a new site) do not alter the equipment that was validated previously
- minor changes to cycle parameters (for example, humidity, pressure, elevation) do not affect the delivered dose and/or sterilant residuals, set out in the approved process and validation
Using a sterilization method that does not adhere to recognized standards may have a significant impact. Consult:
For advice on how to assess the significance of any changes you wish to make to novel sterilization methods, contact the Medical Devices Directorate at meddevices-instrumentsmed@hc-sc.gc.ca.
Note: A change in facility that introduces a new, more challenging organism or bioburden level to the sterilization process which has not been validated before would be significant. This is the case even if the cycle parameters are the same.
Changes to sterile barrier packaging
Packaging changes should also be assessed for their impact on sterilization. In general, any change in packaging characteristics (such as in materials, size, shape, seal width) or configuration (such as in the outer packaging, loading density) is likely significant if it could affect the:
- absorption or penetration of the sterilant
- residual levels (where applicable)
- effectiveness of the sterilization process
- safety of the sterile device
- package seal strength and parameters
Issues of compatibility between the packaging material and the sterilization process must also be taken into consideration.
Changes to previously approved cycles or configurations
A change to the sterilization method or packaging of a sterile medical device is likely not significant if:
- it has already been reviewed and approved in a previous application for a similar device and
- all of the following are true:
- the device is not more difficult to sterilize than the previously authorized device
- the interactions of the device with the proposed packaging do not present a greater risk related to the integrity of the sterile barrier
- the devices are of identical material and similar design (no new worst case design features)
- the proposed changes have been completely represented and approved in a previous new or significant change application
Table of examples
| Device | Proposed change | Significant or not significant |
|---|---|---|
Sterile medical devices |
Sterilization method changed from ethylene oxide (EO) to gamma radiation. |
Significant |
Sterile medical devices |
Biological indicator changed to parametric release. |
Significant |
Sterile medical devices |
Contract sterilizers changed (with no change to cycle parameters) but the method of validating the process is the same. |
Not significant |
Sterile medical devices |
Changes that reduce the SAL. |
Significant |
Sterile medical devices |
Pre-blended EO sterilant changed to EO post-blended with nitrogen where the ultimate concentration of EO in the sterilizer is the same in both cycles, with no change to the critical cycle parameters (delivered dose of EO). |
Not significant |
Sterile medical devices |
Change from using air (mixture of 80% nitrogen and 20% oxygen) to pure nitrogen in the aeration process to avoid explosive gas mixtures, with no change to the critical cycle parameters (delivered dose of EO). |
Not significant |
Sterile medical devices |
Air-flow or heating, ventilating and air conditioning (HVAC) system in the manufacturing environment changed, with the sterilization facility physically and environmentally segregated from the manufacturing line and the device bioburden is not increased above previous level. |
Not significant |
Sterile medical devices |
New alternate EO sterilization facility added with a proposed cycle that is identical to the currently authorized cycle in all critical parameters, but some parameters such as relative humidity are adjusted due to local elevation differences. The new cycle was successfully validated using the overkill method outlined in ISO 11135. |
Not significant |
Sterile medical devices |
Irradiation dose auditing method changed from VDmax to method 1A/1B, method 2 (per ISO 11137) or vice versa |
Significant |
Sterile medical devices with single pouch packaging |
Packaging of a sterile device changed from a single pouch to a new double pouch. |
Significant |
Sterile medical devices with double pouch packaging |
Double sterile barrier packaging replaced with single sterile barrier packaging. |
Significant |
Sterile medical devices |
A proposed packaging change previously reviewed and approved for a similar device sterilized using the same cycle, where the subject device presents no greater challenge to sterilization than the comparable device and the package materials are identical. |
Not significant |
Sterile medical devices |
Packaging dimensions changed or protective enclosure added within the layers of sterile packaging (for example, resin). |
Significant |
Sterile medical devices |
Concentration or exposure time of EO reduced and successfully validated using a method defined in ISO 11135. |
Significant |
Changes to software
Software can be embedded within a medical device or an accessory to a medical device or exist as software as a medical device (SaMD). Software is updated easily and frequently, and thus may undergo several changes during its lifecycle. Health Canada considers some of these changes to be significant.
Examples of significant software changes are those that:
- affect the function or performance specifications associated with the intended use of the device or the intended use of a compatible device whose function is controlled by the software
- introduce a new risk or modify an existing risk that could result in significant harm, including any unintended consequences
- create or necessitate a new risk control measure or a modification of an existing risk control measure for a hazardous situation that could result in significant harm
Examples of non-significant changes if none of the previous changes apply are those that:
- are made solely to return the system into specification of the most recently authorized version of the device
- incorporate a change to the operating system in which the software is running, where the underlying software code is not changed
- are made solely to strengthen cybersecurity and do not have any other impact on the software or the device
- only introduce non-therapeutic and/or non-diagnostic features such as printing, faxing, improved image clarity, reporting format or additional language support
- only modify the appearance of the user interface with little risk of affecting the usability, diagnosis or therapy delivered to the patient
- disable a feature that does not affect the safety or effectiveness of the device
As these are only examples, we recommend that you evaluate additional software changes that can, if not controlled properly, create unexpected software behaviour. Whether a change is significant or not depends on their potential impact on safety or effectiveness.
Other examples of additional software changes that may or may not be significant:
- a re-write in another programming language
- driver modifications
- operating system changes
- not significant if no changes are made to the application
- significant if driver changes are required or the kernel between operating systems is different
- for example, a change in operating system family from Windows to Linux or a major Windows change from XP to Windows 10
Medical devices that use machine learning (ML), in part or in whole, to achieve their intended medical purpose are known as machine learning-enabled medical devices (MLMD). A predetermined change control plan (PCCP) provides a mechanism to address cases where pre-authorization of planned significant changes is needed to address a known risk. This approach can be beneficial in managing certain known risks with MLMD, such as ML system performance degradation over time due to ML model drift.
For medical devices that have been authorized with a PCCP, subsequent changes made according to the authorized PCCP are not significant. Thus, they do not require the submission of a licence amendment application.
For amendments to a device that are outside of an authorized PCCP, including changes to the PCCP itself, consult:
- the Medical Devices Regulations
- other relevant guidance documents in addition to this one
For more information on submission requirements for MLMDs and PCCPs, consult:
Common software change types and unintended consequences
Software changes can come in several different forms, such as:
- infrastructure changes
- for example, changing compilers, programming languages, software drivers
- architectural changes
- for example, new operating system compatibility, software changes to support modified or new hardware
- algorithm changes
- cosmetic changes
- for example, new logos, user interface fonts, colours
- refactoring
- for example, improving efficiency, structure or maintainability
- re-engineering
- for example, reconstituting the software in a new form, replacing aging software
Cosmetic changes are often non-significant. However, the changes listed here may be deemed significant, as there may be unintended consequences when executing software code in often complex software environments. For example, an operating system (OS) upgrade may:
- trigger unintended effects through the use of different drivers or kernels
- require that other software components be updated so they are compatible
You should consider the consequences of the changes to assess whether the change is significant.
A software change is likely not significant if:
- a risk assessment supports this
- it can be verified using the same testing activities that were reviewed by Health Canada for the previously authorized software version and
- does not require software validation testing
Table of examples
| Device | Proposed change | Significant or not significant |
|---|---|---|
Software – skin cancer detection and characterization |
A mobile medical app intended for skin cancer detection and characterization is enhanced by updating its model parameters. There are plans to distribute the upgraded app automatically to existing users. The change affects the sensitivity and specificity of the detection algorithm. The modification has direct impact on the clinical functionality of the device. |
Significant |
Software – IVD device |
A software modification allows an IVD device to improve sample throughput. Modifications include changes to decrease assay times by allowing for shorter sample reaction times. The shorter incubation time may affect diagnostic performance. |
Significant |
Software – chest X-ray application |
A software application using AI-based image analysis of chest X-rays adds a model for detecting endotracheal tubes. The new diagnostic feature introduces a new risk. |
Significant |
Software – diagnostic ultrasound |
A diagnostic ultrasound system has multiple available measurement parameters. Based on a marketing survey of current customers, 1 measurement parameter not directly related to the intended use is removed in a software update. Its removal does not introduce a new risk or modify an existing risk. |
Not significant |
Software – infusion pump |
An infusion pump has 2 occlusion detection alarms: occlusion downstream and occlusion upstream. The software is modified to allow the user to optionally disable 1 alarm. This means the user now has the option to use 1 or both occlusion alarms. This is a change to existing risk controls in the device. |
Significant |
Software – IVD analyzer |
Records in an IVD analyzer are written into a database table. Certain conditions can cause new data to be merged with existing data in the table, which can lead to an incorrect result. The bug is caused by a misworded software requirement leading to a code error. The requirement was rewritten, code was modified and a new database was created for administrative records to prevent merging. The code was rewritten and a new database was added, causing a change in software specifications. |
Significant |
Software – operating system |
An ultrasound system’s operating system has been upgraded to Windows 10 from Windows 7. No changes are made to any of the available software packages, no code changes were made, and both risk analyses and verification testing showed no unexpected results. The operating system belongs to the same OS family and no code changes have been made to make the software compatible with the new OS. Risk analyses and verification testing showed no unexpected results. |
Not significant |
Software – operating system |
A CT scanner’s operating system has been upgraded to Windows 10 from Windows 7. Small infrastructure and architectural changes were made to the software to allow for compatibility. The new OS belongs to the same OS family. However, the change could have unintended consequences that could affect the device’s performance. |
Significant |
Software – operating system |
A blood glucose monitor has been modified to make Android and iOS devices compatible (including control of the device). Previously it had been compatible only with Windows PCs. While the underlying functionality of the device has not changed, a complete rewrite of the algorithms for communication with a new family of operating systems requires full testing and risk analysis. Also, risk controls may be very different in a mobile operating system. |
Significant |
Software – cybersecurity |
A security vulnerability is found in a device’s software through routine cybersecurity monitoring. The software is modified to eliminate the vulnerability. No other changes are made. There is no further impact to the software or device. The cybersecurity vulnerability is eliminated without changing the device’s functionality. Risk analyses determine there is no negative impact to the software or device. |
Not significant |
Software – cybersecurity |
A connection attempt limit is added to an implanted cardiac pacing device that locks access to the device following a set number of failed connection attempts. This is done to prevent unauthorized access. The change is made to strengthen the cybersecurity of the device. However, it may restrict legitimate connections to the device that are clinically important. This change also impacts the device’s safety risk assessment. |
Significant |
Changes in materials for non in vitro diagnostic devices
A change in material type or formulation may affect the chemical, physical and/or electromagnetic properties of the material. A material change may also influence the device’s:
- manufacturing process
- for example, different equipment is required
- design specifications
- for example, device mechanical performance is altered
- biological safety and biocompatibility
- for example, a new potentially harmful leachable
- stability
- for example, device shelf life is reduced
- sterilization method
- for example, ethylene oxide sterilization to replace gamma irradiation is required
- labelling
- for example, warning for allergic reaction to device material is added
Changes related to materials may be intentional or unintentional. Changes in a manufacturing process, equipment or material storage conditions may also affect the properties of device materials.
Examples:
- E-beam/gamma sterilization dosage change may affect the material properties of a crosslinked polymer.
- Material extrusion equipment change may affect the material surface roughness.
- Changes to material reuse/recycling processes or storage conditions for raw material intended for additive manufacturing may affect the properties or performance of the finished device.
All changes should be assessed separately.
The impact of a material change on the safety and/or effectiveness of the device also depends on:
- how the device is to be used
- if the device is invasive or non-invasive
- the type and duration of contact with body tissues or fluids
- if the altered material was used in an authorized device with the same intended use
Note: Please refer to other sections of this guidance document for a material change related to changes in:
- manufacturing process
- facility
- equipment
- control procedure
- design specifications
- for example, device mechanical performance
- sterilization process
- labelling
- compatible Class II devices
- for example, a material change to an independently authorized Class II surgical stapler that’s compatible with authorized Class III surgical staples
The following flowchart and a detailed explanation outline the assessment you must undertake to determine if your change is significant.
Figure 1: Flowchart outlining the decision-making process for changes in material for non-IVDD devices
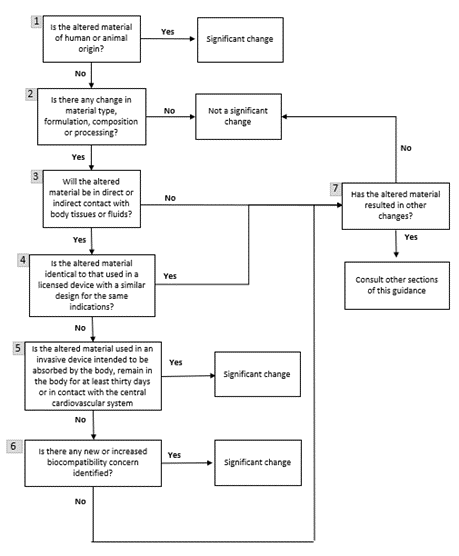
Text description
Figure 1 is a flowchart containing 7 boxes with yes or no questions. Each answer advances the reader through the decision-making process.
Box 1 in the flowchart shows that a change to the sourcing or processing of materials of human or animal origin is significant. For more information, consult:
Box 2 concerns changes to the vendor or supplier. This type of change is not significant if it does not alter the material type, formulation, chemical composition or processing.
Starting from Box 3, the considerations relate to changes that altered the material type, formulation, chemical composition or processing.
First, as indicated in Box 3, you need to determine if the altered material will be in direct or indirect contact with body tissues or fluids.
- Direct contact refers to physical contact of a material with body tissues or fluids.
- Indirect contact refers to a material through which a fluid or gas passes before the fluid or gas comes into physical contact with body tissues or fluids. In other words, the material itself does not physically contact body tissues or fluids.
However, in terms of device safety, there is no difference between direct and indirect contact with body tissues or fluids. A change that involves either type of contact could be significant, pending further considerations.
The change is not significant if the altered material does not have direct or indirect contact with body tissues or fluids and does not cause other changes that could be significant.
If the altered material is in contact with body tissues or fluids, then Box 4 asks if the altered material is used in an authorized device with a similar design for the same indications by the same manufacturer. The change is not significant if the 2 devices are manufactured by the same process and the material is used in the same physical configuration for the same indication in the 2 devices. In addition, there must have been no concerns about the biocompatibility of the authorized device.
The authorized device must have all of the following:
- same or greater risk classification
- same or riskier category of contact
- for example, contact with blood is riskier than contact with intact skin
- same or longer duration of contact
Note: A material change may affect how it interacts with adjacent materials that bond together. Introducing a material used previously in an authorized device to a device where the interactions with adjacent materials are different is a significant change.
If the device doesn't meet those conditions, then Box 5 asks if the altered material is used in an invasive device that is to:
- be absorbed by the body
- remain in the body for at least 30 days or
- be in contact with the central cardiovascular system
If a device meets these conditions, it is a significant change, unless:
- it is a colourant change using an altered dye that complies with List 7 of the FDA's Colour Additive Status List and
- there are no other changes
If the device doesn't meet those conditions, then Box 6 asks about biocompatibility concerns. A material change associated with a new or increased biocompatibility concern is a significant change:
- A new biocompatibility concern refers to the scenario where a new type of biocompatibility testing is required for the device with the altered material.
- An increased biocompatibility concern refers to the scenario where the original biocompatibility testing for the device no longer applies to the device with the altered material.
For example, if an altered material introduces a new leachable that needs to be tested:
- a new biocompatibility concern would result if the leachable is suspected to be genotoxic and no previous genotoxicity testing of the original device was conducted
- an increased biocompatibility concern would result if sensitization testing of the original device is not applicable to the sensitization potential of the leachable
Finally, according to Box 7, if a material change results in other changes such as device specifications, performance, manufacturing process or labelling, consult other sections of this guidance where appropriate.
Table of examples
| Device | Proposed change | Significant or not significant |
|---|---|---|
Hip implant |
Change to the supplier of titanium alloy used to manufacture the hip stem. The material continues to conform with ASTM F136. |
Not significant |
Peripherally inserted central catheter (PICC) |
Colourant change to the insertion hub of a PICC that is part of the fluid path for fluid administration or withdrawal from a patient. The colourant complies with the FDA List 7 Color additives exempt from certification (unless otherwise indicated) and permanently listed for use in medical devices. |
Not significant |
Peripherally inserted central catheter (PICC) |
Colourant change to the flush port of a PICC. The flush port is an access port for flush syringes for IV line clearance or volume block and is not to be used for fluid administration or withdrawal from a patient. The colourant does not come in contact with body tissues or fluids directly or indirectly. |
Not significant |
Cardiovascular |
Polyether block amide (PEBA) changed to polyether ether ketone (PEEK) in a component in a cardiovascular catheter that comes in contact with the cardiovascular system. The same material has been used in another authorized cardiovascular catheter by the same manufacturer. |
Significant |
Dental composite |
Change to the concentration of a dental composite component. The device performance specifications are the same. The device stays in the body for more than 30 days. |
Significant |
Dermal filler |
Change to the crosslinking degree of hyaluronic acid used in a dermal filler. The formulation of the device is the same. The device is absorbed by and stays in the body for more than 30 days. |
Significant |
Breast implant |
Change to a processing equipment, which alters the specifications of the material used in a breast implant. The device stays in the body for more than 30 days. |
Significant |
Hemodialysis system/console |
Change to the material used in the pressure sensor of a hemodialysis system fluid path. The altered material is identical to material used in a temperature sensor in another authorized hemodialysis system fluid path. Both hemodialysis systems are made by the same manufacturer with the same indications for use. They only differ in software features. The material does not contact body fluids for more than 30 days. There is no other concomitant change. |
Not significant |
Oxygenator |
Change to the membrane material used in the gas-exchange part of an oxygenator. The material has not been used in another authorized oxygenator. It is not in contact with body fluids for more than 30 days. However, there is an increased biocompatibility concern about this material. |
Significant |
Suture needle |
Material used in a suture needle changed from 304 Stainless Steel to 316 Stainless Steel. The device is not absorbed by and does not stay in the body for more than 30 days. The manufacturer has another needle authorized that uses the same 316 Stainless Steel. The needle mechanical performance specifications are the same. |
Not significant |
Percutaneous transluminal angioplasty (PTA) balloon dilatation catheter |
Ink used on the proximal marker of a PTA balloon dilatation catheter changed from white hot stamp ink to a black TP300 N50 ink. This ink is in contact with patients but has been used in a authorized similar PTA device by the same manufacturer. The device continues to meet finished product design specifications. The device is not absorbed by and does not stay in the body for more than 30 days. |
Not significant |
PTA balloon dilatation catheter |
Material used in the hypotube of a PTA balloon dilatation catheter changed from 304L Stainless Steel to 304 Stainless Steel. The 304 Stainless Steel hypotube has been used in another authorized PTA catheter by the same manufacturer for the same indications. The device continues to meet the device design specifications. The device is not absorbed by and does not stay in the body for more than 30 days. |
Not significant |
Cardiovascular stent delivery system |
Change to the material of the handle of a cardiovascular stent. There is no change to the design of the handle and the handle does not come in contact with the patient. |
Not significant |
Changes in materials for in vitro diagnostic devices
There is a distinction between in vitro diagnostic devices (IVDDs) and other devices regarding material changes.
Material changes of an IVDD include those made to:
- critical components
- such as antigens, antibodies, primers, conjugates
- other reagents
- such as buffers
- materials that are in direct contact with device reagents
- such as cassettes, solution bottles
Examples of material changes that are likely significant are changes:
- to a critical component of an IVDD
- for which a risk assessment identifies a potential impact on the safety or effectiveness of the device, such as:
- impacts a performance claim
- require a new stability study
- to materials in an IVDD that affect the operating principle of the device
- for example, from immunofluorescence to ELISA
- changes in the operating principle can affect an IVDD’s performance characteristics, including specificity or sensitivity (the impact of each change on the safety and effectiveness of the device should be considered)
- for example, changes in reaction components or materials such as calibration materials that result in altered performance characteristics and are reflected in the labelling are likely significant
Changes to the materials of an IVDD that cause a change to the design, manufacturing process, equipment, control procedures and/or labelling, including stability, are addressed in other sections of this guidance.
Table of examples
| Device | Proposed change | Significant or not significant |
|---|---|---|
IVDD test kit |
Change in the preservative of control material for which internal verification testing showed no change in performance characteristics, including stability. Also, no new hazards have been identified. |
Not significant |
IVDD test kit |
Sodium azide added as a preservative to a reagent of the kit. This introduces a new hazard and requires that a warning be added to the labelling. |
Significant |
IVDD test kit |
Change in PBS buffer within a test kit due to a supplier change with no change to the manufacturer’s specifications. Risk assessment and verification do not identify an impact on safety or effectiveness of the device and no new stability studies are warranted. |
Not significant |
IVDD test kit |
Glass reagent bottles replaced with plastic reagent bottles to address breakage issues, resulting in manufacturer specifications changing for the container. A new stability study is required to establish stability of the reagent using the new container material. |
Significant |
IVDD test kit |
Sample preparation changed to include a stabilizer. A risk assessment shows that adding a stabilizer can impact the assay’s performance. |
Significant |
IVDD test kit |
Change to the sample extraction buffer pH is made to improve extraction efficiency. Risk assessment did not identify an impact on safety and verification activities did not identify an impact on effectiveness. No changes to labelling are made. |
Not significant |
IVDD test kit |
External positive control source material changed from human antibody to non-human antibody. The change means a new stability study as the stability of the newly formulated control has not been established. |
Significant |
IVDD test kit |
Primary packaging changed from a less water-permeable to a more water-permeable material (for example, highly impermeable foil to plastic). Risk assessment and verification activities identify an impact on device stability. |
Significant |
Changes to labelling
Labelling includes:
- user manuals
- patient labelling
- instructions for use
- labels affixed on the device
- other provided documents that include information about using the device
Labelling changes are often triggered by:
- changing user requirements
- changes to a device, including changes to performance specifications and materials
You must consider each labelling change separately, as well as the total impact of the labelling changes over time.
Changes that trigger a labelling change should also be assessed against all other relevant sections of this guidance.
Changes concerning indications for use, intended use and clinical benefits
Changes to the intended use or indications for use are significant unless they are within the current scope of indications set out in the labelling. This follows the general principles where:
- interpolation is less likely to result in a significant change
- extrapolation is more likely to result in a significant change
A significant change would be to add a new subgroup of patients to the indications or intended use. This group may be considered higher risk than the more general population group identified in the previous indications or intended use.
However, this change is likely not significant if the identified subgroup of patients was added for clarification and would have been covered within the indications for use for the authorized device.
Example of a significant change:
- the change includes pediatric patients, but the previous indications associated with the labelling for the authorized device were general and did not refer to patient age
Example of a non-significant change:
- a device was indicated for use in patients who experienced a heart attack and
- a change was made to add “adult” patients who experienced a heart attack to the indications for use and
- the device was a well-established technology where early intervention is not associated with increased long-term or life-time risks
In general, reducing indications for use to a subset of the original population is a non-significant change.
Despite this, the following would be considered a significant change:
- a limitation to the indications for use is introduced because of concerns associated with the safe or effective use of the device
- note that a contraindication or a warning should also be added to the labelling in this case
- wording is modified to imply that more clinical evidence exists to support the claim beyond what was previously submitted and reviewed by Health Canada
- involves new wording and new claims
- the indications for use are modified to allow for use only in cases where a medical condition exists, such as hypertension above 150 mm Hg
- requires clinical evidence to support the claim
Examples of other significant changes to labelling:
- changes that alter label content about when, where or by whom the device is used
- new claims associated with earlier interventions compared to current labelling claims for the authorized device
- new claims that may change the environment in which the device is used
- such as from a hospital setting to an office or home setting
- new claims that alter who can use the device
- such as from a health care practitioner to a technician, patient or caregiver
Examples of changes to warnings and precautions that may not be significant:
- minor changes clarifying the existing wording of warnings and precautions for a device
- removing a warning or precaution where the same information is included and presented identically elsewhere in the labelling
- adding a warning or precaution not driven by new information pertaining to the safety and effectiveness of the device
Examples of changes to warnings and precautions that are significant:
- adding or removing a contraindication
- removing a warning or precaution unless equivalent information continues to be included elsewhere in the labelling in an equivalent manner
- adding a warning or precaution that is driven by new information on the safety and effectiveness of the device
- removing possible adverse events if listing these events is part of a required risk mitigation
Adding a clinical benefit to labelling may introduce new claims or expand existing claims made in the indications for use. This is likely to be a significant change in most cases.
Examples of a significant change:
- new claims (explicit or implied) of safety and of effectiveness that need to be supported with data
- new representation of uncertainties about performance that require supporting data
- for example, stating that a total knee replacement has 80% reliability at 15 years
Example of a non-significant change:
- adding a clinical benefit where the statements are fully included within the scope of the authorized indications for use and the supportive information is in the existing authorized labelling
New labelling may be introduced that targets different populations (such as patient-specific labelling) or is created for marketing purposes.
Examples of a non-significant change:
- new labelling does not introduce new claims or statements about device safety, effectiveness, uncertainty or therapeutic mechanisms, that are beyond the scope of indications for use, intended use or the general description of use included in the labelling for the currently authorized device
- labelling changes that clarify instructions to make the device easier, safer or more effective to use
For IVDDs, the following are examples of changes that are likely significant:
- identifying a new subpopulation for which clinical data have not previously been provided
- The identification of this new subpopulation would not be a significant change if clinical data have already been provided and reviewed for the subpopulation.
- adding or removing limitations not captured in the intended use but that appear elsewhere in the package insert
- This change is non-significant if it does not impact the correct use, performance or interpretation of correct results (supported by usability assessment or clinical experience).
- removing a warning or a precaution
- Adding a warning or precaution is likely not a significant change unless it also impacts the intended use.
Changes concerning reprocessing, sterilizing, cleaning or disinfection
Medical devices that are reprocessed, sterilized, cleaned or disinfected by the end user must include manufacturer validated guidance on the cleaning, disinfection and/or sterilization process.
Consult:
Changes to the recommended sterilizing, cleaning or disinfecting method or product as previously submitted and reviewed in relation to an authorized class III or IV device may be deemed significant. Removing a recommended method or product from the labelling or package insert may not be a significant change if alternate, validated options authorized in Canada are still included in the labelling.
Changes concerning other regulatory jurisdictions
For information on adding a clinical benefit to labelling, please refer to the subsection on Changes concerning indications for use, intended use and clinical benefits. Including additional languages required in other regulatory jurisdictions is not a significant change.
Changes related to references in the labelling are significant if they:
- include articles that relate specifically to off-label use of the medical device in relation to the currently authorized labelling
- add references related to off-label usage
- add new claims to the labelling associated with referenced materials if the claims have not been previously reviewed and incorporated into labelling authorized by Health Canada
When it comes to references, the following are not significant changes:
- introducing new references to data housed in third-party databases that may be required for other regulatory approvals if this database indicates the data are available for non-Canadian audiences
- including references to publicly available information, such as journal articles
You should also remove references to obsolete devices from the instructions for use once a device is no longer available for sale and use in Canada. This is not considered a significant change.
Changes concerning the useful life of a product
The following are examples of a significant change:
- introducing a new statement about the projected useful life of a device that adds a specific timeframe (for example, 90% freedom from device failure at 3 years) which has not been included in the currently authorized labelling
- making claims for shelf-life extension if the:
- protocols and methods for determining shelf life have been changed or were not reviewed in a previous application
- results or acceptance criteria are significantly different from previously reviewed data
- reducing shelf life if a change has affected the safety or effectiveness of the device or other considerations have changed the risk/benefit ratio
Examples of a non-significant change:
- adding broad statements without specific claims, such as:
- knee implants have a finite life and future revision or replacement may be required
- the projected useful life depends on use and patient characteristics
- adding statements associated with simulated bench testing that demonstrate a minimum life based on non-clinical testing models, with a clear disclaimer that patient factors may increase or decrease the expected lifetime of the device if Health Canada has previously reviewed the data
- This would be significant if the simulated bench testing had not previously been submitted to Health Canada as part of the submission supporting the authorization.
- making claims for shelf-life extension, including for IVDDs, if the new claims were validated by you, with similar findings based on using the same protocols and methods as the initial validation submitted to Health Canada at the time of authorization (the same protocol including the same acceptance criteria previously accepted by Health Canada)
- If any of the parameters change during the validation of the shelf-life extensions, the change becomes significant.
Changes concerning compatible devices
You should clearly communicate within the labelling what devices are available in Canada. Information should clearly indicate how the device can be used safely and effectively with authorized compatible devices that are available in Canada. Be sure to remove references to obsolete devices from the instructions for use once a device is no longer available for sale and use in Canada (not a significant change).
Adding references in the instructions for use to unauthorized devices or to devices that were previously authorized but are no longer available in Canada due to obsolescence are not significant changes as long as:
- the labelling does not include language that is promotional or is being used to advertise the unauthorized device for sale
- there is no claimed interoperability between the unauthorized device and your device that is not substantiated, for example:
- labelling for a proposed bone plate references unauthorized screws and you have evidence the device is safe and effective if used with those screws, then it is acceptable to include a reference to the unauthorized screws
- non-significant change
- labelling for a patient monitor references an unauthorized diagnostic probe manufactured by another manufacturer for which you do not have evidence of safety and effectiveness
- significant change, as this would require evidence be submitted to demonstrate safety and effectiveness
- labelling for a proposed bone plate references unauthorized screws and you have evidence the device is safe and effective if used with those screws, then it is acceptable to include a reference to the unauthorized screws
- the primary indication for use does not depend on this unauthorized device
- for example, whether the unauthorized device is mentioned in the indications for use
- use of the unauthorized device is not required for your device to meet the requirements of sections 10 to 20 of the regulations
- use of the unauthorized device is not required for your device to meet the requirements of paragraph 68.11(2)(g) of the regulations
- including the reference to unauthorized devices is not misleading to the user, as the instructions for use for the unauthorized device may be ignored and still allow the device to be used safely and effectively
Adding compatibility claims to labelling of a Class III or IV device with a separately authorized Class III or IV device is often a significant change. This applies to both of the following situations:
- adding the claim to both Class III and IV devices, where both devices are from the same manufacturer
- adding the claim to the Class III or IV device, where the manufacturer of the second device is different
- for example, adding a compatibility claim between a polyethylene liner and a separately authorized femoral head
Adding a compatibility claim with an authorized device that’s similar to a currently listed compatible device is not necessarily a significant change.
Adding a compatibility claim would not be significant if:
- the differences between the proposed and current compatible device are considered non-significant according to this guidance and
- the currently listed compatible device and the new proposed compatible device (which is also authorized in Canada) are manufactured by the same manufacturer
However, adding a new compatible device is likely significant if:
- the manufacturer of the new proposed compatible device has not previously been listed as a manufacturer of a compatible device
- the new compatible device impacts the indications for use of either device
If you made a compatibility claim with a device from a different manufacturer, you should monitor for possible changes that would render the compatibility invalid. A significant change made by a third-party manufacturer to a compatible device could impact the safety and effectiveness of your device. This would be a significant change.
For changes in compatibility claims associated with a compatible Class II device, refer to the following section on Class III or IV amendments concerning separate compatible Class II devices.
Removing an approved compatibility claim may be considered a significant change if:
- the removal is due to concerns with the safe or effective use of the assembled compatible devices and
- the concerns are about how the devices interact with each other and how the system will perform as a result
Changes concerning magnetic resonance
In general, a change to the magnetic resonance (MR) safety claim of an authorized medical device, in terms of scan conditions under which a scan may be safely performed, is significant.
A non-significant change would be where:
- an implantable device contains no ferro-magnetic or para-magnetic materials (for example, a polymer implant) and new MR labelling claims are being added
- MR conditional claims are reduced to a subset of previously authorized conditions
Table of examples
| Device | Proposed change | Significant or not significant |
|---|---|---|
All devices |
Deleting a contraindication (such as "not for pediatric use"), deleting a contraindication against lip augmentation for a dermal filler or removing a contraindication against the use of a dental implant in patients who smoke. |
Significant |
All devices |
Labelling change to include additional languages, other than French or English, required in other regulatory jurisdictions. |
Not significant |
Dermal filler |
Deleting potential adverse events, such as granuloma formation. |
Significant |
Percutaneous aortic valve |
Introducing an additional warning that the device could embolize if not deployed completely and confirmed under fluoroscopy. |
Not significant |
Stent graft |
Modifying the indications for use to exclude femoral implantation, but this was previously indicated. This change is implemented due to safety concerns identified in post-market use. |
Significant |
Radiofrequency generator |
The radiofrequency generator is approved for use with authorized radiofrequency probes for the indication of creating radiofrequency lesions in nervous tissue. Adding another mode to the generator to be used with other authorized radiofrequency probes that are approved for use in the intervertebral disc to coagulate and decompress disc material. |
Significant |
Radiofrequency probe |
The radiofrequency probe is indicated for ablating nervous tissue (used peripherally). Modifying indications for use so that the probe may now be used in the central nervous system (for example, brain). |
Significant |
Structural heart defect device (PFO closure) |
Modifying indications for use to clarify that it is to be used in adults older than 21 years of age. This is the patient population that would normally be considered within the scope of a general indication for use. |
Not significant |
Ventricular assist device |
Modifying indications for use from an adult population to a population with a mass greater than 20 kg. |
Significant |
Total knee replacement |
Adding clinical benefit to state that a total knee replacement has 80% reliability at 15 years. |
Significant |
Class III and IV amendments for compatible Class II devices
“Compatibility” is the ability of a device, when used with 1 or more other devices, to achieve the overall clinical purpose without the user having to modify or adapt any part of the combined devices.
An important requirement in demonstrating safety and effectiveness of medical devices that are to be used together is compliance with section 18 of the Medical Devices Regulations. Under section 18, a medical device intended to be used together with other medical devices must:
- be compatible with every other medical device with which it interacts and
- not adversely affect the performance of the medical devices used together
This requirement applies to compatible devices that reside on the same authorization and those that are authorized separately.
Refer to the following notice for the situation when the devices are authorized separately:
When a change is made to 1 or more of the compatible devices, you must ensure that the combination of medical devices continues to meet section 18 requirements. As well, as per item (d) of the definition of “significant change”, a change to the intended use of the device includes new or extended use.
This section of this guidance applies to the following 2 scenarios:
- the labelling of a Class III or Class IV device is being changed to indicate compatibility with a Class II device, even if the formal “intended use and/or indications for use” statement stays the same
- a change is being made to an authorized Class II device that is already indicated as compatible with an authorized Class III or IV device
To evaluate whether either of these 2 scenarios are significant changes for the Class III or IV authorization, you must assess if they may affect the safety or effectiveness of the Class III or IV device. Consider the following factors:
- Critical nature of the Class II device
- the more critical the Class II device is to overall system function (including safety or effectiveness), the more likely the compatibility change could affect the safety or effectiveness of the Class III or IV device
- the following would typically be significant changes:
- longer duration of use
- new clinical indications beyond or more specific compared to those on the Class III or IV claims
- different patient population
- change that necessitates new clinical studies
- change to the therapy delivered
- new operating workflow or user interface
- Differences in key design specifications between the Class II device(s), which are previously indicated as compatible with the device, and the new or modified Class II device
You should identify and analyze how such differences could impact safety and effectiveness of the overall system. This includes assessing whether the change may affect such factors as compatibility, performance and risk mitigations.
Table of examples
| Device | Proposed change | Significant or not significant |
|---|---|---|
Class II swab used to collect endocervical specimens |
The validation of the swab for use with a specific Class III Chlamydia test was reviewed as part of the application for that test. Change made to the specifications of the swab (for example, in material or design) that could impact the safety and/or performance of the test. |
Significant |
Class II administration set |
Adding a new Class II administration set to a Class III programmable infusion pump’s list of compatible administration sets. Following an engineering analysis and risk assessment, the manufacturer determined that the design parameters of the new administration set fall within the design parameters of the administration sets. These are already indicated as compatible with the pump (for example, tubing dimensions and material, number and characteristics of integrated components, manufacturing processes, including sterilization). Verification testing was done to verify that the pump performs as expected with the new administration set using the same testing activities that were reviewed in a previous application. The testing did not produce any unexpected results. There are no changes to the intended use or indications for use compared to the authorized administration sets. |
Not significant |
Class II insulin infusion set |
Adding a new Class II insulin infusion set to a Class III or IV programmable insulin pump’s list of compatible infusion sets. The new infusion set is indicated for a longer wear period than the infusion sets that are previously indicated as compatible with the pump and longer than stated in the pump labelling. The longer wear period necessitated new testing activities to support the safety and effectiveness of the therapy delivered by the overall system. |
Significant |
Class II delivery catheter indicated for use in the delivery of transcatheter aortic valves |
A Class IV transcatheter aortic valve is authorized as compatible with a Class II delivery catheter. In this example, the Class II device’s labelling specifies the authorized access routes, but the Class IV device’s labelling does not. Introducing a design change to the Class II delivery catheter system allows for a different access route than the one authorized in the initial application. The design changes to the delivery catheter impact the clinical performance of the devices when they are used together, including a potential increase in risk and complications in delivering the transcatheter aortic valve. New clinical testing was required to support the safety and effectiveness of the overall system. |
Significant |
Change in reprocessing of a Class III or IV device |
An authorized Class III or IV device is labelled for use with a Class II disinfecting device. A change in the specifications of the Class II disinfecting device could impact the performance or safety of the Class III or IV device when it is disinfected using the Class II device. |
Significant |
Class II surgical stapler handle |
Change to an authorized Class II surgical stapler handle, which could impact the performance of the compatible Class III stapler reloads. |
Significant |
Class II sound processor |
Change to the software of a sound processor authorized separately as a Class II device, but compatible with a Class III cochlear implant and/or a Class IV brain stem implant, as well as Class II hearing aids. The change in software could impact the performance of the Class III or IV systems or safety mitigations. |
Significant |
Changes to diagnostic ultrasound systems
Health Canada recognizes there are a number of diagnostic ultrasound systems authorized in Canada that have an established safety profile in Canada and in other regulatory jurisdictions. Consequently, a change made to a diagnostic ultrasound system is not significant if all of the following apply:
- intended use of the system has not changed and modifications do not introduce intracardiac or intravascular imaging through catheter-based transducers
- modifications do not introduce sterile use or sterilization where previously not indicated and do not affect previously indicated sterile use
- modes of operation for the modified device are well established
- well established modes: A-mode, B-mode, M-mode, Doppler (CW, Color, PW, Power, Combination), speckle-tracking, tissue harmonic imaging and combination
- not well established modes: shear wave elastography, acoustic attenuation mapping, transmission based imaging, sound speed measurement
- modifications do not lead to acoustic outputs that exceed the recommended maximum acoustic output levels (consult Device licence applications for ultrasound diagnostic systems and transducers)
- ISPTA.3 < 720 mW/cm2 for most applications, <430 mW/cm2 for cardiac, <17 mW/cm2 for ophthalmic
- ISPPA.3 < 190 W/cm2 for most applications, <28 W/cm2 for ophthalmic
- MI < 1.9 for most applications, <0.23 for ophthalmic
- modifications do not result in any ultrasound interrogation parameters outside each of the following ranges:
- 1 to 20 MHz centre frequency
- 0 to 7 MPa peak rarefactional pressure
- 1 to 100 cycles in a pulse (except for CW Doppler)
- 100 to 20,000 Hz pulse repetition frequency
- modifications do not use novel mechanical or thermal effects for imaging or measurements
- measurements and analyses are clearly described and the user can adjust associated control parameters
- image processing should be reversible or the original image should be available to the user
- user is able to edit or adjust user-activated post-processing applications used for measurements (for example, segmentation and registration)
- where possible, user should be able to edit assumed values, parameters or thresholds in equations or algorithms used to generate additional outputs based on measurements of anatomical dimensions, tissue velocity or pixel intensity
- labelling provides complete information about the processing or compression algorithms used by the device, when appropriate
- integrated transducer element check is performed each time a transducer is connected to the main system or activated to help ensure appropriate transducer performance
- for example, an impedance check of each element may provide a preliminary evaluation of the element integrity and function
- transducer surface temperature falls within the requirements of IEC 60601-2-37
- if the device is for endocavity use, labelling includes validated cleaning/disinfecting instructions and identifies appropriate probe covers
Table of examples
| Device | Proposed change | Significant or not significant |
|---|---|---|
Diagnostic ultrasound system |
Adding M-mode imaging to the modes of operation of the device. |
Not significant |
Diagnostic ultrasound system |
Adding a new transducer with the same indications for use as the system’s. The acoustic output and interrogation parameters are within the defined limits. |
Not significant |
Diagnostic ultrasound system |
Adding a software feature that uses shear-wave elastography to measure liver stiffness. |
Significant |
Page details
- Date modified: