
Reporting adverse reactions to marketed health products: Draft guidance document for industry
Download in PDF format
(1.35 MB, 47 pages)
Effective Date: Month dd, yyyy
Forward
Guidance documents are meant to provide assistance on how to comply with governing statutes and regulations. Guidance documents also provide assistance to staff on how Health Canada mandates and objectives should be implemented in a manner that is fair, consistent and effective.
Guidance documents are administrative instruments not having force of law and, as such, allow for flexibility in approach. Alternate approaches to the principles and practices described in this document may be acceptable provided they are supported by adequate justification. Alternate approaches should be discussed in advance with the relevant program area to avoid the possible finding that applicable statutory or regulatory requirements have not been met.
As a corollary to the above, it is equally important to note that Health Canada reserves the right to request information or material, or define conditions not specifically described in this document, in order to allow the Department to adequately assess the safety, efficacy or quality of a therapeutic product. Health Canada is committed to ensuring that such requests are justifiable and that decisions are clearly documented.
This document should be read in conjunction with relevant sections of other applicable guidance documents.
On this page
- 1 Introduction
- 2 General Procedures for Expedited Adverse Reaction Reporting
- 3 Good Case Management Practices
- 4 Types of Adverse Reaction Reports
- Appendix 1 Glossary: Definitions and Terminology
- Appendix 2 References
- Appendix 3 Abbreviations
- Appendix 4 Contact Information
- Appendix 5 Other Adverse Reaction Reporting Programs Outside the Scope of this Document
- Appendix 6 World Health Organization Causality Algorithm
- Appendix 7 Summary of Expedited Post-Market AR Reporting Requirements to MHPD
- Appendix 8 Determining when to report cases found in the Canada Vigilance Adverse Reaction Online Database
1 Introduction
1.1 Scope
This guidance document provides market authorization holders (MAHs) (the entity that holds the Drug Identification Number (DIN), Natural Product Number (NPN) or Homeopathic Medicine Number (DIN-HM)) with assistance on how to comply with the Food and Drug Regulations, and the Natural Health Products Regulations with respect to reporting adverse reactions (ARs) for marketed health products.
ARs for marketed health products within the scope of this guidance document are to be reported to the Canada Vigilance Program of the Marketed Health Products Directorate (MHPD) of Health Canada. This guidance document covers the collection of individual AR reports by MHPD for the following marketed health products:
- pharmaceutical drugs (which includes prescription and non-prescription pharmaceutical drugs);
- biologics as set out in Schedule D to the Food and Drugs Act (which include biotechnology products, DIN-assigned manufactured blood products and vaccines);
- radiopharmaceutical drugs set out in Schedule C to the Food and Drugs Act; and
- natural health products as defined in Section 1 of the Natural Health Products Regulations.
In addition to the requirement for MAHs to submit AR reports in accordance with the Food and Drug Regulations and the Natural Health Products Regulations (collectively these two sets of regulations are referred to hereafter as "the Regulations"), Health Canada has powers to request additional information on ARs as set out in the RegulationsFootnote 1Footnote 2Footnote 3.
For further information on AR reporting for health products not covered by this guidance document, please refer to their respective guidance documents:
- Blood and blood components: Guidance Document: Blood RegulationsFootnote 23
- Cells, tissues, and organs: Guidance Document for Cell, Tissue and Organ Establishments - Safety of Human Cells, Tissues and Organs for TransplantationFootnote 24
- Sperm and ova: Guidance Document: Interpretation of the proposed regulations under the Assisted Human Reproduction ActFootnote 25
- Medical devices: Guidance Document for Mandatory Problem Reporting for Medical DevicesFootnote 26
Note that drugs and natural health products authorized for phases I-III clinical trials involving human subjects pursuant to Part C, Division 5 of the Food and Drug RegulationsFootnote 4 and Part 4 of the Natural Health Products Regulations, respectively, are not within the scope of this guidance document. Only spontaneous and solicited ARs associated with a suspect product, marketed in Canada, that is administered to a subject outside a study protocol (phase I-III CTA), must be reported to MHPD in accordance with Divisions 1 and 8 of the Food and Drug Regulations or Section 24 of the Natural Health Products Regulations (see section 4.2.2).
While pharmaceutical products that contain ingredients derived from cannabis (DIN-assigned products) are within the scope of this guidance document, cannabis itself is not, regardless if it is used medically or recreationally (for further information on AR reporting of cannabis by licence holders, see the Cannabis Act and Cannabis Regulations).
Additionally, drugs authorized for sale under the Special Access Programme, the issuance of an Interim Order by the Minister of Health, and Access to Drugs in Exceptional Circumstances, are also not within the scope of this guidance document.
This guidance document does not cover the preparation and collection of summary reports, such as annual summary reports (ASR) and issue-related summary reports. For assistance on how to comply with the Food and Drug Regulations and the Natural Health Products Regulations with respect to annual (C.01.018) and issue-related summary reports (C.01.019), MAHs should refer to the guidance document Preparing and Submitting Summary Reports for Marketed Drugs and Natural Health Products.
For adverse reaction reporting outside the scope of this document, Appendix 5 provides further details on these other reporting programs.
1.2 Adverse Reaction Reporting by Market Authorization Holders
Every MAH is required to report serious ARs known to them involving their marketed health products in accordance with the requirements of the Food and Drugs Act and the Regulations. The success of Health Canada's AR reporting system depends on the quality, completeness, accuracy, and timeliness of the information submitted. Reporting of ARs and the monitoring thereof remain a viable means of identifying previously unrecognized, rare or serious ARs. This may result in updating product safety information, facilitating decisions on regulatory actions such as withdrawal of a product from the Canadian market, contributing to international data regarding risks and effectiveness of health products, and imparting health product safety knowledge that benefits all Canadians.
In facilitating reporting of ARs by MAHs, Health Canada has harmonized to the greatest extent possible the recommendations in the International Council on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) guidance documents:
- Clinical Safety Data Management: Definitions and Standards for Expedited ReportingFootnote 5 (ICH E2A)
- Maintenance of the ICH Guideline on Clinical Safety Data Management: Data Elements for Transmission of Individual Case Safety ReportsFootnote 6 (ICH E2B(R2))
- Periodic Benefit-Risk Evaluation ReportFootnote 7 (ICH E2C(R2))
- Post-approval Safety Data Management: Definitions and Standards for Expedited ReportingFootnote 8 (ICH E2D)
- Pharmacovigilance PlanningFootnote 9 (ICH E2E)
- Report of the Council for International Organizations of Medical Sciences (CIOMS) V Working Group: Current Challenges in Pharmacovigilance: Pragmatic ApproachesFootnote 10
- MedDRA Term Selection: Points to consider
The MAH, in accordance with subsections C.01.016 – C.01-020 Part C Division 1 and subsection C.08.007 – C.08.008 of Part C Division 8 of the Food and Drug Regulation is expected to continually monitor their health products, reports ARs, and conduct all post-market monitoring of their products for the duration of their products' market authorization. As such, there is no specified period for a MAH's reporting of ARs or post-market monitoring activities of their products. Given the lifecycle approach to health product regulation, there is continual learning and monitoring regarding the quality, safety and effectiveness of health products.
1.3 Distinguishing Between Adverse Reactions and Adverse Events
This guidance document applies to expedited reporting of adverse reactions (ARs) rather than adverse events (AEs). ARs to marketed health products covered by this document may be generated from unsolicited and solicited reports.
An AR is characterized by the fact that a causal relationship between the drug and the occurrence is suspected. The definition of adverse reactionFootnote 1Footnote 11 (see Appendix 1) implies that there is a suspected relatedness to the administered health product. Health professionals and consumers report adverse reactions because of their suspicion of the relatedness of an adverse event to a health product. The description of experiences in these reports should therefore be considered adverse reactions. Reportable ARs also include those suspected of being the result of drug interactions (e.g., drug-drug interactions, drug-natural health product interactions, drug-food interactions).
An adverse event, as defined in ICH E2DFootnote 8, means any untoward medical occurrence in a patient administered a medicinal product and which does not necessarily have to have a causal relationship with this treatment. An AE can therefore be any unfavourable and unintended sign (for example, an abnormal laboratory finding), symptom, or disease temporally associated with the use of a medicinal product, whether or not considered related to this medicinal product.
1.4 Serious Adverse Reaction Reports
A serious adverse reaction is defined at C.01.001 in the Regulations as a noxious and unintended response to a drug or natural health product that occurs at any dose and that requires in-patient hospitalization or prolongation of existing hospitalization, causes congenital malformation, results in persistent or significant disability or incapacity, is life-threatening or results in death.
Medical and scientific judgement by a qualified health professional should be exercised in deciding whether expedited reporting is appropriate in other situations, such as medically important events that may not be immediately life-threatening or result in death or hospitalization but may jeopardize the patient or may require intervention to prevent one of the other outcomes listed in the definition from the Regulations. Health Canada asks that these medically important cases be reported on an expedited basis as well. Examples of medically important events are intensive treatment in an emergency room or at home for allergic bronchospasm, blood dyscrasias or convulsions that do not result in hospitalization, development of drug dependency or drug abuse.
The seriousness criterion selected by the adverse reaction reporter should not be downgraded from serious to non-serious if the receiver (e.g., MAH) disagrees with the seriousness reported by the reporter.
1.5 Determining if an Adverse Reaction is Unexpected
An AR is considered unexpected when its nature (i.e., specificity or outcome), severity or frequency is either not identified, or is not consistent with the terms or description used in the Canadian product labelling such as the product monograph, labelling standards, information approved for market authorization, or the product label. In cases where the MAH is uncertain whether an AR is expected or unexpected, the AR should be treated as unexpected.
For cases that involve a fatal outcome, AR reports should be considered unexpected unless the Canadian product labelling specifically states that the AR may be associated with a fatal outcome.
CProduct class ARs should not automatically be considered expected for the subject health product. Product class ARs should be considered expected for the suspected health product only if described as specifically occurring with the product in the Canadian product labelling as illustrated in the following examples:
- "As with other health products of this class, the following undesirable effect occurs with Product X."
- "Health products of this class, including Product X, can cause..."
If the AR has not been documented with Product X, statements such as the following are likely to appear in the Canadian product labelling:
- "Other health products of this class are reported to cause..."
- "Health products of this class are reported to cause..., but no reports have been received to date with Product X."
In these situations, the AR should not be considered as expected for Product X.
1.6 Regulations Pertaining to Adverse Reaction Reporting
The sections of the applicable regulations that set out the AR reporting requirements are listed below.
- Prohibition (C.01.016)
- Serious Adverse Drug Reaction Reporting (C.01.017)
- Annual Summary Report and Case Reports (C.01.018)
- Issue-related Summary Report (C.01.019)
- Maintenance of Records (C.01.020)
- New Drugs (C.08.007(1)(h), C.08.008(c))
Annual Summary Report and Case Reports (C.01.018) and Issue-related Summary Reports (C.01.019) are not covered under this guidance document but are discussed in the guidance document Preparing and Submitting Summary Reports for Marketed Drugs and Natural Health Products – Guidance Document for Industry
Natural Health Products Regulations
- Reaction Reporting (Section 24)
2 General Procedures for Expedited Adverse Reaction Reporting
Every MAH should put into place written procedures for the receipt, evaluation, and reporting of ARs.
RElectronic reporting of ARs is the preferred and most reliable method for MAHs to comply with regulatory timelines. As soon as they are capable of doing so, MAHs should enrol with Health Canada as trading partners to submit individual case safety reports (ICSRs) electronically in accordance with the technical requirements and business (validation) rules set out in Health Canada's Trading Partner Management Office (TPMO) document which is available upon request (see Appendix 4 for contact information). Electronic reporting of ARs significantly reduces the amount of time and effort involved in the reporting process.
MAHs that do not yet meet the technical requirements to submit ARs electronically, as defined in the aforementioned instruction document, may continue to send AR reports to MHPD by fax or by mail (see Appendix 4 for contact information). The preferred reporting format for AR reporting by MAHs via fax and mail is as follows:
- for drugs, the Council for International Organizations of Medical Sciences (CIOMS) Form IFootnote 13
- for drugs and natural health products, the Mandatory Adverse Reaction Reporting Form for IndustryFootnote 14
Health Canada does not consider the transmission of AR reports via email a secure method of submission. As such, AR reports should not be sent to Health Canada via email; instead, electronic, fax or mail submission of AR reports is recommended.
For more information on the expectations with respect to pharmacovigilance systems, MAHs should consult Health Canada's Good Pharmacovigilance Practices (GVP) Guidelines GUI-0102Footnote 15.
2.1 Domestic and Foreign Adverse Reaction Reports
For drugs, MAHs must submit domestic and foreign AR reports to MHPD pursuant to Part C, Division 1 (C.01.016 C.01.017) and for new drugs must submit reports of unusual failure in efficacy pursuant to Part C, Division 8 (C.08.007, C.08.008) of the Food and Drug Regulations once their drugs are available for sale in Canada. These reporting obligations (see sections 2.1.1 and 2.1.2) for MAHs commence when the MAH sells a drug, which can occur for example when a MAH offers a drug for sale, exposes a drug for sale or has a drug in its possession for sale and distribution.
For natural health products, MAHs must submit domestic and foreign AR reports to MHPD as set out in Section 24 of the Natural Health Products Regulations once their health product is licensed to be marketed in Canada.
To facilitate the processing of AR reports, the MAH should indicate if the report is domestic or foreign by clearly indicating the country where the reaction occurred.
The regulatory reporting time clock starts on the day when the MAH first has all of the information that satisfies the minimum criteria for an AR report (see Section 3.1). This date should be considered day 0. If the collection of ARs is performed by a separate entity through a contractual agreement (e.g. co-marketer or third-party company), the day on which the contracted person or organization receives the ARs, should be considered day 0. Please refer to section 3.6 for more information on contractual agreements.
2.1.1 Domestic Adverse Reaction Reports
AR reports concerning reactions occurring in Canada to a product that is marketed in Canada are considered "domestic" AR reports.
In order to report in compliance with the Regulations, the MAH should report to MHPD, within 15 calendar days of receiving the minimum information required to satisfy the minimum criteria for an AR report (Section 3.1), the following solicited and unsolicited domestic reports:
- serious ARs (expected and unexpected)
- an unusual failure in efficacy for new drugs (see Appendix 1 for New Drug definition)
Inquiries regarding new drug status for health products marketed in Canada should be referred to the appropriate Directorate (i.e., Biologics and
2.1.2 Foreign Adverse Reaction Reports
Foreign AR reports are those concerning reactions occurring outside Canada to a product that is marketed in Canada.
In order to report in compliance with the Regulations, the MAH should report to MHPD, within 15 calendar days of receiving the minimum information required to satisfy the minimum criteria for an AR report (Section 3.1), the following solicited and unsolicited foreign reports:
- serious unexpected ARs
Unexpectedness is determined by the absence of an AR in relevant Canadian labelling such as the product monograph, labelling standards, information approved for market authorization, or the product label.
All foreign serious unexpected AR reports involving the MAH's and international counterpart's foreign products with the same combination of active ingredients irrespective of variations in the formulation, dosage form, strength, route of administration, or indication, that is also marketed in Canada must be reported to MHPD in accordance with the Regulations (e.g., a MAH that sells a marketed health product in Canada with active ingredients X, Y, and Z, must report all foreign serious unexpected AR reports involving their foreign products with the same combination of active ingredients X, Y, Z).
If the product source, brand, or trade name is not specified, the MAH should assume that it was its own product, although the report should indicate that the specific brand was not identified.
2.1.2.1 Canada's Access to Medicines Regime
In response to public health problems afflicting many developing and least-developed countries, Canada passed an Act to amend the Patent Act and the Food and Drugs Act (The Jean Chrétien Pledge to Africa). The Act, which came into force on May 14, 2005, creates a legislative framework that enables manufacturers to obtain an authorization (i.e., compulsory licence) allowing them to make, construct and use a patented invention solely for the purpose of exporting a pharmaceutical product to eligible importing countries. The provisions of the Act are now incorporated in the Patent Act and the Food and Drugs Act.
Compulsory licence holders are subject to the requirements for reporting foreign adverse reactions to health products sold under Canada's Access to Medicines Regime (CAMR). Compulsory licence holders submitting these reports to MHPD are requested to specify the following on the cover sheet: FOREIGN ADVERSE REACTION, CANADA's ACCESS TO MEDICINES REGIME.
2.2 Other Adverse Reaction Report Types
Sections 2.2.1 to 2.2.3 apply to all products that fall under the Regulations:
2.2.1 Overdose, Medication Error or Occupational Exposure
Cases of overdose, medication error or occupational exposure associated with serious ARs are subject to expedited reporting (within 15 days) in accordance with the Regulations. As with all ARs, routine follow ups should be performed to ensure that the information is as complete as possible with regard to symptoms, treatment, outcome, and context of occurrence (e.g., error in prescription, administration, dispensing, dosage, etc.). The MAH should collect any available information on these cases related to its products.
2.2.2 Pregnancy Exposure
MAHs are expected to follow up all pregnancy reports from health professionals and consumers where the embryo/foetus could have been exposed to one of its health products. For consumer reports, it is appropriate for the MAH to seek permission from the patient to only follow up with their health professional. The MAH must apply all principles outlined in this guidance document and the Regulations pertaining to reporting requirements, including determination of seriousness and minimal criteria for submitting an AR report.. Reports of pregnancy exposure with no associated adverse reactions should not be reported as ARs. When an active substance, or one of its metabolites, has a long half-life, this should be taken into account when considering whether a foetus could have been exposed (e.g., if health products taken before the gestational period should be considered). Care should be taken when reporting ARs related to the embryo/foetus that the patient and the parent/child relationship are accurately identified in the report, and that the AR information is attributed to the correct patient. For example, if an AR occurs in both the parent and foetus, then two separate AR reports should be submitted, if the reporting requirements are met.
2.2.3 Discontinued Products
In accordance with the Regulations, the MAH must report any AR information received prior to the discontinuation of sale in Canada. Although the MAH is not obliged to report any new cases of adverse reactions received following the product's discontinuation, Health Canada may request the provision of this information. If a serious AR was known to the MAH before the discontinuation of sale, they must still report as per the expedited reporting requirements even if the end of the 15-day reporting timeframe as required by the Regulations is after the date on which sales were discontinued. Follow-up information for cases known to the MAH prior to the discontinuation of sale should be reported to MHPD in accordance with the Regulations, and should be sought as part of the follow-up practices described under Section 3.4.
When expired and unexpired lots of a discontinued product continue to be available in pharmacies, the MAH is still under obligation to report ARs to MHPD if this information was received by the MAH prior to the discontinuance. As mentioned above, Health Canada may still request the MAH to provide information that it receives following the discontinuation of sales.
2.2.4 Products with DINs Reported as Dormant
In accordance with section C.01.014.71 of the Food and Drug Regulations, a manufacturer must notify Health Canada within 30 calendar days after a market notified product has not been sold on the Canadian market for a period of 12 consecutive months. Upon notification, the status of this product is updated as dormant on Health Canada's online Drug Product Database.
The MAH must continue to report AR information received for these dormant products. Products with a dormant status meet the definition of "sell" as outlined in the Food and Drugs Act and therefore continue to be subject to the conditions of sale set out of the Regulations. This includes, but is not limited to, the reporting of ARs with respect to the product. Additional information on products with DINs that have been reported as dormant can be found in the Guidance Document: Regulatory requirements for Drug Identification Numbers (DINs).
Subsequent to notifying Health Canada of the 12-month period without sale, if the MAH determines that they will not resume sale of the product on the Canadian market as per C.01.014.72, they must submit a sale discontinuation notification as outlined in Section 6.2 of the aforementioned guidance document. The MAH will not be required by the Regulations to report any new adverse reactions following the product's discontinuation, although Health Canada may request the provision of this information. The reporting of adverse reactions received after discontinuation is still highly encouraged.
2.2.5 Unusual Failure in Efficacy (only applies to new drugs)
The MAH must report an unusual failure in efficacy of a new drug in accordance with subsections C.08.007, C.08.008 of Part C, Division 8 of the Food and Drug Regulations. A new drug is defined as a product that has been given a notice of compliance as per subsection C.08.002 of Part C, Division 8 of the Food and Drug Regulations. Please note that natural health products are not subject to this requirement unless the Natural Health Product Regulations refer to the Food and Drug Regulations.
For new drugs marketed in Canada, domestic reports of unusual failure in efficacy must be reported in accordance with C.08.007(h) to MHPD within 15 calendar days of the receipt of information by the MAH. Inquiries regarding new drug status for health products marketed in Canada should be referred to the appropriate Directorate (i.e., Biologics and Radiopharmaceutical Drugs Directorate or Therapeutic Products Directorate).
The unusual failure in efficacy refers to a new drug that fails to produce the expected intended effect, despite being used as per the Product Monograph. The underlying principle is that if a health product fails to produce the expected intended effect, there may be an adverse outcome for the patient, including an exacerbation of the condition for which the health product is being used. An unusual failure in efficacy should be reported to MHPD in an expedited fashion regardless of whether the event itself is imminently serious. Clinical judgement should be exercised by a qualified health professional from the MAH to determine if the problem reported is related to the product itself, rather than one of treatment selection or disease progression since health products cannot be expected to be effective in 100% of the patients.
One example of unusual failure in efficacy is a previously well-stabilized condition that deteriorates when the patient changes to a different brand or receives a new prescription. Another example of a case that should be reported on an expedited basis is a life-threatening infection where the failure in efficacy seems to be due to the development of a newly resistant strain of bacterium previously regarded as susceptible.
In cases where the MAH is uncertain whether an AR should be considered as a report of unusual failure in efficacy, the AR should be treated as such and submitted to MHPD accordingly.
3 Good Case Management Practices
3.1 Minimum Criteria for an Adverse Reaction Report
Complete information for the final description and evaluation of an AR report may not be available within the time frame required for reporting. Nevertheless, for regulatory purposes, AR reports must be submitted within the prescribed time, as long as the following minimum criteria are met:
- (a) An identifiable reporter (source)
- (b) An identifiable patient
- (c) A suspect product
- (d) An adverse reaction.
Ideally, more comprehensive information would be available on all cases from the outset, but in practice MAHs will often have to follow up with the reporter after initially submitting the report to seek additional information. Follow-up AR reports should be clearly documented as such (see section 3.4). The MAH is expected to exercise due diligence to collect any key data elements (see Section 3.8) that are lacking at the time of initially submitting the report. The MAH should provide all information that is available and relevant in the initial AR report and not just that which satisfies the minimum criteria. See section 3.8 for a list of key data elements which enhance report quality.
It is important that at the time of the original report, sufficient details about the patient and reporter be collected and retained to enable follow-up in accordance with the collection, use and disclosure provisions of the Personal Information Protection and Electronic Documents Act or equivalent provincial privacy legislation.
3.2 Assessing Patient and Reporter Identifiability
Patient and reporter identifiability is important to avoid case duplication, and facilitate follow-up of appropriate cases. The term "identifiable" in this context refers to the verification of the existence of a patient and a reporter. AR cases without specific identifiers (e.g., reporter name or patient gender) may meet the first two reporting criteria outlined in section 3.1, however, follow-up information should be actively sought and submitted as it becomes available. All parties submitting case information or approached for case information should be identifiable: not only the initial reporter (the initial contact for the case), but also others supplying information. In addition, in the event of second-hand reports, every reasonable effort should be made to verify the existence of an identifiable patient and reporter.
For MAHs submitting AR reports manually, one or more of the following should automatically qualify a patient as identifiable: age or age category (e.g., adolescent, adult, elderly), gender, patient identification number, or reference to "a patient". For MAHs submitting AR reports electronically, at least one element within the Patient Characteristics block is mandatory. If patient information is unknown to the sender or cannot be transmitted due to privacy laws, patient initials should be populated with "UNKNOWN" or "PRIVACY", respectively.
In the absence of qualifying descriptors (e.g., age, gender), a report referring to a number of patients should not be regarded as a case until the minimum four criteria for case reporting are met. For example, "a few patients experienced" should be followed up for patient-identifiable information before reporting to MHPD. The four minimum criteria must be met for each reported patient and an individual report should be submitted for each identifiable patient. The regulatory time clock (i.e. day 0) does not begin until all four minimum criteria are met, e.g. once "a few patients" are identified as patients X, Y, and Z, and the other three criteria are met.
Provide as many patient identifiers in appropriate structured fields when reporting. For instance, do not simply include "female" as a patient identifier when it is known that the patient was a 29-year-old female. Inclusion solely in the narrative field is not sufficient.
3.3 The Role of Narratives
The objective of the narrative is to summarize all relevant clinical and related information, including patient characteristics, therapy dates, medical history, clinical course of the event(s), diagnosis, and AR(s) including the outcome, laboratory evidence (including normal ranges), and any other information that supports or refutes an AR (e.g., rechallenge information). The narrative should serve as a comprehensive, stand-alone "medical story". Care should be taken by the MAH to ensure that the information in the narrative (e.g., patient identifiers, ARs, indication, and medical conditions) is accurately captured in the appropriate data fields.
Abbreviations and acronyms should be avoided, with the possible exception of laboratory parameters and units. Key information from supplementary records including summarized relevant autopsy or post-mortem findings should be included in the report, and their availability should be mentioned in the narrative, identified in the appropriate tags if submitting electronically and supplied on request. Clinical judgement should be exercised by a qualified health professional from the MAH to determine what information should be submitted. Personal identifiers should only be submitted in accordance with the collection, use and disclosure provisions of the Personal Information Protection and Electronic Documents Act or equivalent provincial privacy legislation.
3.4 Follow-up Information
Follow-up information should be actively sought and significant new information must be submitted by the MAH as it becomes available for appropriate amendment to the database and files in MHPD. Follow-up AR reports should be appropriately linked to the initial report. For reporting and case management purposes, the initial report is considered to be the first report that is sent to MHPD. Follow-up information should be clearly identified within the report, and should be updated in the narrative sequentially by the date it was received by the MAH. Corresponding data fields should be updated accordingly. The MAH should ensure that the MAH received date of any follow-up information does not precede the latest received date of the previous report version.
When additional medically significant information is received for a previously reported case, the reporting time clock (see Section 2.1) is considered to begin again for submission of the follow-up report. Significant follow-up information received by the MAH for serious domestic ARs and serious unexpected foreign ARs must be reported to MHPD within 15 calendar days. For the purpose of reporting, significant follow-up information relates to, for example, new suspected adverse reaction(s), additional or changed suspect product, a change in the causality assessment and any new or updated information on the case that impacts its medical interpretation. Therefore, the identification of significant new information requiring expedited reporting always necessitates the qualified health professional's medical judgement. Routine tests conducted independently of the adverse reaction(s), or follow-up results for a test previously reported may not require expedited reporting unless the results are deemed significant (e.g., impact the medical interpretation of the case) by the qualified health professional from the MAH.
In addition, a case initially classified as a non-expedited report, would qualify for expedited reporting upon receipt of follow-up information that indicates the case should be re-classified (e.g., from non-serious to serious). In the reverse scenario, where an initially serious case is re-classified as non-serious, this information is still subject to the 15-day expedited reporting timeline; thereafter additional information for these re-classified cases is not subject to 15-day timelines, in so long as the case remains non-serious.
In any scheme to optimize the value of follow-up, the first consideration should be prioritization of case reports by importance. The priority for follow-up should be as follows: cases which are (1) serious and unexpected, (2) serious and expected, and (3) non-serious and unexpected. Although non-serious and unexpected cases are not expedited, MAHs are encouraged to pursue follow-up information on these reports. In addition, cases of "special interest" also deserve extra attention as high priority (e.g., ARs under enhanced or active surveillance at the request of Health Canada), as well as any cases that might lead to a labelling change decision.
Follow-up information should be obtained from the reporter by the MAH, via a telephone call and/or site visit and/or a written request. The MAH should ask specific questions it would like to have answered. Follow-up methods should be tailored towards optimizing the collection of missing information. If appropriate, written confirmation of details given verbally should be obtained. All attempts to obtain follow-up information (whether or not successful) should be documented as part of the case file, particularly on the serious cases. The number of follow-up attempts along with the date and time of each should be documented to reflect sufficient diligence.
To facilitate the capture of clinically relevant and complete information, use of a targeted questionnaire/specific form is encouraged, preferably at the time of the initial report. Ideally, qualified health professionals should be involved in the collection and the direct follow-up of reported cases. For serious ARs, it is important to continue follow-up and report new information until the outcome has been established or the condition is stabilized. The amount of time devoted to follow up such cases is a matter of the qualified health professional's judgement.
3.5 Evaluation and Coding of Adverse Reaction Reports
The purpose of careful medical review by qualified health professionals is to ensure correct coding and evaluation of medical information. Preferably, information about the case should be collected from the health professionals who are directly involved in the patient's care. Regardless of the source of an AR report, the MAH should carefully review the report for the quality and completeness of the medical information. The review should include, but is not limited to, the following considerations:
- Has a diagnosis been assigned?
- Have the relevant diagnostic procedures been performed?
- Were alternative causes of the reaction(s) considered?
- What additional information is needed?
The Medical Dictionary for Regulatory Activities (MedDRA), an ICH initiative (ICH M1), is an internationally accepted, clinically validated medical terminology developed to share regulatory information about medical products used by humans. MedDRA provides a set of terms which consistently categorizes medical information and is meant to standardize the terminology through which medical regulatory information is classified, stored, retrieved, presented and communicated.
In order to avoid loss or distortion of communicated information, it is recommended that MedDRA be used as a standard for the coding of medical information in AR reports. MedDRA coding should be applied to all medical information (e.g. ARs, medical history, and indications) whenever possible. For trading partners who are submitting ICSRs electronically to MHPD, exclusive use of the current version of MedDRA is required.
When using the MedDRA terminology for the coding of AR reports, two supporting ICH-endorsed guides are available for MedDRA users:
- MedDRA Term Selection: Points to Consider (MTS:PTC)Footnote 16 document for accurate and consistent term selection, and
- MedDRA Data Retrieval: Points to Consider (MDR:PTC)Footnote 17 for consistent use of MedDRA for data analysis/output and presentation of medically meaningful review and analysis of clinical data.
The MedDRA Points to Consider documents aid in standardising the use of MedDRA between various users. Term selection methods and quality assurance procedures for coding adverse reaction reports should be documented in MAH-specific coding guidelines which should be based on, and not in conflict with, the MTS:PTC. In some cases, where there is more than one option for selecting terms, the MTS:PTC document identifies a preferred option. A MAH should be consistent in the option that they chose, and should document their selected option in their MAH-specific coding guidelines.
Every effort should be made to use AR terms consistently and in accordance with recommended standards for diagnosis. The report should include the verbatim term as used by the reporter, or an accurate translation of it if provided in a language other than English or French. Any MAH personnel receiving reports should provide an unbiased and unfiltered report of the information from the reporter. While the report recipient is encouraged to actively query the reporter to elicit the most complete account possible, inferences and imputations should be avoided in report submission. However, clearly identified evaluations by the MAH are considered appropriate when they state that they reflect the MAH opinion and not that of the reporter.
When a case is reported by a consumer, his/her description of the event should be retained, although confirmatory or additional information from any relevant qualified health professionals should also be sought and included as part of the follow-up practices described under Section 3.4.
As described in Sections 1.4, 4.1.1, and 4.2, where the receiver (e.g., MAH) disagrees with the reporter's seriousness criterion or suspicion of a causal relationship between the suspected health product and the reported adverse reaction, these should not be downgraded by the MAH. The opinions of both the reporter and the MAH should be recorded in the AR report.
3.6 Contractual Agreements
The marketing of many health products increasingly takes place through contractual agreements between two or more companies or within the same company, which may market the same product in the same or different countries or regions. Arrangements vary considerably with respect to inter-MAH communication and regulatory responsibilities. Therefore, it is essential that explicit licensing or contractual agreements specify the processes by which an exchange of safety information, including timelines and regulatory reporting responsibilities, are taking place. Pharmacovigilance personnel should be involved in the development of any agreements from the beginning. Processes should be in place to avoid duplicate reporting to the regulatory authority (e.g., assigning the responsibility to one MAH for literature screening).
Whatever the nature of the arrangement, the MAH is ultimately responsible for regulatory reporting. Therefore, every effort must be made between the contracting partners to minimize the data exchange period so as to promote compliance with MAH reporting responsibilities. For MAHs that have a contractual agreement for the initial collection of ARs, the regulatory timeclock begins when the contracted person or organization first receives the AR and the four minimum criteria, for reportability, an identifiable reporter, an identifiable patient, a suspect product and an adverse reaction are met.
3.7 Records to be Held for Auditing
The Food and Drug Regulations require that records of the AR case reports and summary reports, be maintained. For drugs, the MAH must retain records for 25 years after the day on which they were created as per C.01.020. It is also recommended that these records be easily accessible within 72 hours. The records for unusual failure in efficacy must be retained for 7 years as per C.08.007(1.1).
For natural health products, records of the AR case reports and summary reports should be maintained to permit audit or submission on request. A minimum 25-year retention period is recommended from the date the record was created. It is also recommended that these records be easily accessible within 72 hours.
3.8 Key Data Elements
The following is a list of key data elements that enhance the quality of an AR report. The MAH is expected to exercise due diligence in obtaining information on as many listed items as are pertinent to the case.
- 1. Patient Details
- Unique identifier (to readily locate the case for follow-up purposes; do not use the patient's full name)
- Gender
- Age, age category (e.g., adolescent, adult, elderly)
- Height and weight
- Pre-existing conditions
- Medical history
- Relevant family history
- 2. Suspected Health Product(s)
- Brand name (or proprietary drug name) as reported [the brand name is the name assigned by the MAH, used to distinguish the health product, and under which the health product is sold or advertised, and includes any name extensions or modifiers (prefix or suffix)]
- Canadian authorization numbers such as a Drug Identification Number (DIN); Homeopathic Medicine Number (DIN-HM) and Natural Product Number (NPN) which appear on the label
- Common Name such as the International Nonpropriety Name (INN)
- For natural health products, it is important to include the Latin binomial, author reference, family (genus and species), type of extract (e.g., aqueous versus alcoholic, including percent of solvent), part of the plant used (in the case of an herbal product), ingredients and quantity of each (for homeopathic products, potency of each ingredient). If a particular ingredient in a combination is suspected, this should also be identified.
- Batch/lot number
- Indication(s) for which suspect health product was prescribed or tested, or indicate if unknown
- Dosage form and strength
- Daily dose (specify units, e.g., mg, ml, mg/kg) and regimen
- Route of administration; unknown route of administration should be indicated as such
- Starting date and time
- Stopping date and time, and duration of treatment
- For vaccines, indicate the number of previous doses of each vaccine. For example, if the event occurred after a series of several vaccinations (e.g., 3 doses of hepatitis B vaccine) give details of prior immunizations in the narrative
- 3. Concomitant and Other Treatment(s)
The same information as in item 2 should be provided for the following:
- Concomitant health products (including non-prescription, over-the-counter medicinal products, natural health products, dietary supplements, complementary and alternative therapies, etc.)
- Health products used in the treatment of the AR.
- Any other health products being used at the time of the AR, including medical devices
- 4. Details of AR(s)
- Full description of reaction(s), including body site and severity
- The criterion (or criteria) for regarding the report as serious if reported as such
- Description of the reported signs and symptoms
- Specific diagnosis for the reaction
- Onset date (and time) of reaction
- Stop date (and time) or duration of reaction
- Dechallenge and rechallenge information; unknown action taken should be indicated as such
- Relevant diagnostic test results and laboratory data
- Setting (e.g., hospital, out-patient clinic, home, nursing home)
- Outcome (recovery and any sequelae)
- For a fatal outcome, stated cause of death
- Relevant autopsy or post-mortem findings
- Relatedness of product to reaction(s)/event(s)
- 5. Reporter Details
- Reporter type (consumer, health professional, etc.)
- Profession (specialty)
The following is a list of the administrative and MAH details that should always be included with the report:
- Source of report (e.g., clinical trial, literature, spontaneous, regulatory authority)
- Date the event report was first received by MAH
- Country in which the reaction occurred
- Type (initial or follow-up) and sequence (first, second, etc.) of case information reported to Health Canada
- Name and address of MAH
- Name, address, electronic mail address, telephone number, and facsimile number of contact person of MAH
- MAH's identification number for the case (the same number should be used for the initial and follow-up reports on the same case).
4 Types of Adverse Reaction Reports
4.1 Unsolicited Reports
An unsolicited report is a spontaneous report which is defined by the ICH as an unsolicited communication by a health professional or consumer to a MAH, regulatory authority (e.g., Health Canada) or other organization that describes one or more ARs in a patient who was given one or more health products and that is not derived from a study or any organized data collection scheme. For these types of reports, MAHs can infer implied causality.
4.1.1 Consumer Reports
Consumer AR reports should be handled as spontaneous reports irrespective of any subsequent "medical confirmation". Emphasis should be placed on the quality of the report and not on its source.
If a MAH receives a report from a consumer, the MAH should encourage the patient to report the reaction through their health professional or permission should be sought to contact the consumer's health professional. In addition, the MAH should attempt to obtain as much information as possible from the patient. The description of the experiences in these reports should therefore be considered adverse reactions.
A valid case of suspected adverse reaction initially submitted by a consumer cannot be downgraded to a report of non-related adverse event if the contacted health professional (e.g., nominated by the consumer for follow-up information) disagrees with the consumer's suspicion. In this situation, the opinions of both the consumer and the health professional should be included in the AR report.
If the spontaneous case meets the minimum criteria for reporting, it would require expedited reporting to MHPD in accordance with the Regulations. Given the implied causality for spontaneous cases, causality assessment by the MAH is irrelevant in terms of determining reportability of a case, but nonetheless considered a good practice to include as a key data element to enhance the quality of a report.
Even if the MAH receives reports that do not qualify for expedited regulatory reporting, the cases should be retained as they are applicable to the annual summary reporting requirements in the Regulations.
4.1.2 Scientific Literature Reports
Every MAH is expected to screen the worldwide scientific literature on a regular basis by accessing widely used systematic literature reviews or reference databases. Cases of ARs from the scientific and medical literature, including relevant published abstracts from meetings and draft manuscripts, might qualify for expedited reporting. For the purpose of expedited reporting set out under C.01.017 of the Food and Drug Regulations (FDR), it is reasonable for MAHs who only sell their health products in Canada to limit their literature searches to local journals. However, regardless of whether a MAH sells their health products domestically or internationally, and in line with section C.01.018 of the FDR, global literature searches are expected in the preparation of annual summary reports according to the risk profile of the product and other factors. This is discussed in sections 2.2.3 and 2.2.4 of Preparing and Submitting Summary Reports for Marketed Drugs and Natural Health Products - Guidance Document (please see MHPD contact information in Appendix 4 to obtain more details around these requirements).
An individual report with relevant medical information must be provided for each identifiable patient. The publication reference(s) should be given as the report source. Additionally, the MAH is expected to provide the article to MHPD upon request. All MAH offices (e.g. global and regional offices) should be aware of publications in their local journals and to bring them to the attention of the MAH safety department as appropriate.
For foreign literature reports, all foreign serious unexpected ARs involving the MAH's foreign products with the same combination of active ingredients irrespective of variations in the formulation, dosage form, strength, route of administration, or indication, that is also marketed in Canada must be reported to MHPD in accordance with the Regulations (see Section 2.1.2).
If the product source, brand, or trade name is not specified, the MAH should assume that it was its own product if the MAH has market authorization for that active moiety in the country where the AR occurred, although the report should indicate that the specific brand was not identified.
If multiple products are mentioned in the article, a report should be submitted only by the MAH whose product is suspected. The suspect product(s) is/are those identified as such by the article's author.
In general, it is recommended that the frequency of the literature searches be at least every two weeks. A qualified health professional from the MAH should use their expertise and scientific judgement to determine the appropriate frequency of literature searches as the required level of safety monitoring may vary for well-characterised low risk health products marketed by the MAH. In order to advocate for an alternative schedule for literature searches, MAHs can provide a defensible, objective rationale as to why they should conduct their literature searches on a less frequent basis. MAHs should readily provide their justification upon request and/or during inspections to justify the frequency at which the literature scans are performed for their products. The appropriate timing, frequency and nature of environmental scanning would depend on such factors as the risk profile of the product, any known or specific emergent issues, the scheduling of the summary report, etc. Identified and potential safety issues, as well as knowledge gaps (e.g., toxicity in vulnerable groups, interactions with other products, emergent use patterns) may require more active monitoring.
As such, the following are a few points of consideration with respect to providing more objective justifications of an alternative search frequency:
- Has the MAH been conducting more frequent searches of the published literature and not identified any new safety information?
- If so, how often were the searches conducted and how long has the MAH had this procedure in place?
- What databases/websites were searched and what search parameters were used?
- How does the MAH propose to be able to identify any serious safety issue if one does arise?
- Has the MAH submitted documents which support all of the aforementioned considerations?
The regulatory reporting time clock starts as soon as the MAH has knowledge that the case meets minimum criteria for reportability. The MAH should make reasonable attempts to contact the study author/organization to collect the minimum criteria, assess and report accordingly. Should these attempts be unsuccessful, the MAH should keep a record of their follow-up efforts.
4.1.3 Stimulated Reports
Stimulated reports are those that may have been motivated, prompted or induced and can occur in certain situations, such as notification by a Health Professional Communication (HPC), Health Canada-issued Public Advisory and/or Public Communication (PC), literature report, publication in the press, or questioning of health professionals by MAH representatives. These reports should be considered unsolicited (spontaneous) in nature and must be reported to MHPD in accordance with the Regulations.
4.1.4 Reports via the Internet/social media
MAHs should regularly screen websites under their management or responsibility for potential AR case reports. It is the MAHs' responsibility to establish their own internal processes and procedures for periodic screening and record keeping, for which they can provide a defensible, objective rationale upon request and/or during inspections. MAHs are not expected to screen external websites for AR information. However, if a MAH becomes aware of an AR on a website that it does not manage, the MAH is expected to review the case, follow-up accordingly, and determine whether it should be reported.
MAHs should consider utilising their websites to facilitate AR data collection, e.g., by providing AR forms for reporting or by providing appropriate contact details for direct communication.
Cases from the Internet should be handled as unsolicited reports. For the determination of reportability, the same minimum criteria (i.e., identifiable reporter, identifiable patient, suspect product and AR) should be applied as for cases provided via other ways. If the minimum reporting criteria are met and the case is considered "reportable" and must be forwarded to the MHPD in accordance with the Regulations.
For these reports, the regulatory reporting time clock is considered to start on the day when the MAH first has all of the information that satisfies the minimum criteria for an AR report (see Section 3.1).
4.1.5 Other Unsolicited Reports
If a MAH becomes aware of a case report from non-medical sources (e.g., the lay press or other media), it should be handled as an unsolicited report. Reportability should be determined using the same minimum criteria (i.e., identifiable reporter, identifiable patient, suspect product and AR) as for other reports.
4.2 Solicited Reports
Solicited reports are defined by the ICH as those derived from organized data collection systems, which include clinical trials, registries, post-approval named patient use programs, other patient support and disease management programs, surveys of patients or health professionals, or information gathering on efficacy or patient compliance. AR reports obtained from any of these sources should not be considered unsolicited. Since such reports are regarded as solicited in nature, one cannot infer implied causality, unlike the convention for spontaneous reports. Solicited reports should also not be confused with stimulated reports (see Section 4.1.4).
For the purposes of AR reporting, solicited reports should only be submitted if there is a reasonable possibility that the health product caused the AR as determined by a qualified health professional of the MAH. A "reasonable possibility" means that the relationship cannot be ruled out. For example, using the World Health Organization criteria for causality applicable to AR reporting, any case reports that fall within the criteria of Certain, Probable, Possible, or Unlikely (see Appendix 6) must be reported to MHPD. In any case where an underlying illness or another health product may have contributed to the adverse event, the report should still be considered an AR, as the causality cannot be ruled out. Submitting solicited AR reports where no causality assessment has been conducted will result in "noise" in the database and reduce the efficacy of surveillance activities in determining potential safety signals.
For solicited reports, where the receiver (e.g., MAH) disagrees with the reasonable possibility of causal relationship between the suspected health product and the adverse reaction expressed by the reporter, the case should not be downgraded to a report of non-related adverse event. The opinions of both the reporter and the MAH should be recorded in the AR report.
4.2.1 Reports from Patient Support and Disease Management Programs
A patient support and disease management program is a service associated to the MAH that involves direct interaction with patients and/or patient caregivers which are geared towards helping patients manage medication and/or disease outcomes, understanding their condition and providing advice on managing disease, or providing a service or arranging financial assistance for patients. Examples of these programs include, but are not limited to, telephone services for patients to obtain direct advice, nurse-initiated calls for medicine compliance management, surveys collecting other patient data, and establishment of large patient registries.
Reports generated through these programs are usually considered solicited reports and are reportable in accordance with the Regulations, as these reports are not generated in the usual spontaneous manner that is the premise upon which unsolicited reporting systems are based. Instead, they are usually obtained through a focused line of questioning designed to capture suspected ARs and/or clinically relevant information.
However, it is important to note that reports received through these programs may also be considered spontaneous (unsolicited) if information not actively sought through the MAH's organized data collection scheme is provided.
- For example, reports from these programs should be classified as consumer reports (see section 4.1.1), when the health care professional associated to the MAH's patient support and disease management program is not involved in the care of the patient and thus does not have access to sufficient information to verify the events reported (e.g. access to their medical records).
- Other situations when reports could be classified as spontaneous include financial assistance programs (e.g., co-pay programs), or specialty pharmacy programmes, as long as there is no organised data collection (i.e., when there is no solicited communication), when the reporter reaches out to the firm, and when there is no planned interaction.
When classifying safety information as solicited or spontaneous, it is the MAH's responsibility to provide a defensible, objective rationale for their classification.
| Report type | Did the reporter suspect/confirm a causal relationship between the suspected product and the AR? | As per the MAH's own causality assessment, is there a reasonable possibility that the health product caused the AR? | Is the MAH required to report expeditiously to MHPD? |
|---|---|---|---|
| An unsolicited report is submitted by a reporter (e.g. submitted by a HCP on behalf of their patient or directly by the patient/patient's relative) | Yes | Yes | Yes |
| No | Yes | ||
| No | Yes | Yes | |
| No | No | ||
Notes:
|
|||
Follow-up information should only be submitted on an expedited basis if it is considered to be medically significant by the qualified health professional from the MAH (see Section 3.4).
4.2.2 Reports from Studies
For studies, this section of the guidance document refers to the MAH's post-market AR reporting requirements for marketed health products, Division 1 (C.01.016 and C.01.017) and Division 8 (C.08.007(h) and C.08.008(c)) of the Food and Drug Regulations and Section 24 of the Natural Health Products Regulations.
MAHs are also subject to AR reporting for health products used in phase 1-III clinical trials involving human subjects where the MAH is the sponsor of the study, in accordance with the requirements listed in Part C, Division 5 of the Food and Drug Regulations, or Part 4 of the Natural Health Products Regulations. These requirements are not within the scope of this guidance document (see Appendix 5 for program information).
4.2.2.1 Market Authorization Holder Sponsored Studies
The MAH may sponsor various pre-market (Phases I to III) or post-market studies involving their products. Post-market studies refer to Phase IV studies performed after the drug has been approved by the regulator for the market, and related to the approved indication.
Studies subject to post-market AR reporting requirements (e.g., phase IV studies) should be monitored in a way that ensures that all serious expected and unexpected domestic ARs, serious unexpected foreign ARs and cases of domestic unusual failure in efficacy for new drugs are reported to the MAH by the investigator(s) so that the MAH can provide such reports to MHPD within the 15-day period specified in the Regulations.
Investigators should be provided with the definition of what constitutes a serious AR for reporting purposes. In such cases, it is important to distinguish between "reactions" and "events", not only for administrative purposes but also to minimize the instances of reporting adverse events that are clearly unrelated to therapy. MAHs should help investigators understand their role in assessing the possible relationship between an adverse event and the administration of a health product during post-marketing studies.
Comparator and concomitant products used in these studies are within the scope of this guidance document. It is the sponsor's responsibility to decide whether active comparator and concomitant product adverse reactions should be reported to the MAH of the active control and/or directly to MHPD.
It should be noted that a domestic clinical trial AR report for a marketed product used for the purpose of a MAH-sponsored Phase I-III clinical trial in Canada is subject to Division 5 of the Food and Drug Regulations, and must be reported to the appropriate clinical division (e.g. Therapeutic Products Directorate, or Biologics and Radiopharmaceutical Drugs Directorate). Sponsors reporting adverse reactions to an appropriate clinical division of Health Canada in accordance with Division 5 of the Food and Drug Regulations or Part 4 of the Natural Health Products Regulations should not report those ARs to MHPD in duplicate. Please refer to Appendix 5 for information on the relevant clinical divisions.
4.2.2.2 Non-Market Authorization Holder Sponsored Studies
A MAH may receive study AR reports where its product was a comparator treatment (and therefore used in accordance with approved labelling) or was a product the patient was taking concomitant to the study medication but was suspected of causing an AR. The source of these reports may be another MAH who is sponsoring the study, a private investigator or an academic centre.
The MAH must apply all principles outlined in this guidance document and the Regulations pertaining to reporting requirements, including determination of seriousness, causality, and minimal criteria for submitting an AR report. The MAH should not alter the causality assessment of the trial product(s) provided by the trial sponsor and should include any narrative of the trial sponsor regarding causality, if available. The MAH should assess causality on its own marketed health product(s).
A MAH may offer support such as a drug and/or funding to independent investigators and institutions such as academia, medical research charities or research organizations in the public sector to conduct post-authorization studies within the scope of the NOC or NOC-C (e.g. Investigator Sponsored/Investigator Initiated clinical trials). For such trials, the investigator/institution is considered to be the sponsor of the trial and therefore, must fulfill all the regulatory obligations for expedited AR reporting, unless there is a contractual agreement in place between the MAH and the investigator/institution, delegating the responsibilities to the MAH. The party responsible for expedited reporting is to determine whether active comparator and concomitant product ARs should be reported to the MAH of the active control and/or directly to MHPD.
This guidance document does not cover ARs observed part of post-authorization studies outside of the parameters of the NOC/DIN (e.g. new indication), which is subject to Division 5 of the Food and Drug Regulations, and must be reported to the appropriate clinical division (e.g. Therapeutic Products Directorate, or Biologic and Radiopharmaceutical Drugs Directorate).
4.2.2.3 Post-Study Adverse Reactions
Although such information is not routinely sought or collected by the sponsor, serious adverse reactions that occurred after the patient had completed a clinical study (including any protocol-required post-treatment follow-up) will possibly be reported by an investigator to the sponsor. Such cases should be regarded for expedited reporting purposes as though they were study reports. Therefore, a causality assessment is needed for a decision on whether or not expedited reporting is required.
4.2.3 Blinded Study Reports (in Phase IV)
If the MAH receives a serious domestic AR report or a serious unexpected foreign AR report from the investigator that is blinded to individual patient treatment, the code must be broken before submitting the report to MHPD. Although it is advantageous to retain the blind for all patients prior to final study analysis, it is recommended that, when a serious AR occurs, the MAH seek a third party to break the blind only for that specific patient, even if the investigator has not broken the blind. It is also recommended that, when possible and appropriate, the blind be maintained for individuals such as biometrics personnel, who are responsible for analysis and interpretation of results at the conclusion of the study.
4.3 Reports from Regulatory Authority Sources
As part of their surveillance activities, MAHs should consult Canadian and foreign regulatory authorities to identify AR reports involving their products and determine if these meet the criteria for expedited reporting.
If the product source, brand, or trade name is not specified, the MAH should assume that it was its own product if the MAH has market authorization for that active moiety in the country where the AR occurred, although the report should indicate that the specific brand was not identified (i.e. the suspect product would be the active ingredient(s) in the product and not a brand name). For the purpose of expedited reporting as it relates to C.01.017:
- If MAH is the DIN holder and sells their health products only in Canada, this MAH is only required to report serious domestic ARs to MHPD. The MAH is also encouraged (but not required) to report foreign unexpected ARs related to products with the same combination of active ingredients.
- If MAH is the DIN holder, and sells their health products both in Canada and abroad, this MAH is required to report both serious domestic ARs and serious foreign unexpected ARs to MHPD.
- If MAH is the DIN holder and sells their health products only outside Canada, this MAH is encouraged to report to MHPD domestic serious and/or foreign unexpected serious ARs related to products with the same combination of active ingredients.
In Canada, the Canada Vigilance Adverse Reaction (CV) Online Database is updated on a monthly basis to capture domestic AR reports that were sent directly to the Canada Vigilance Program by health professionals, consumers or other MAHs. As of December 16, 2019, new authorities provided through the Protecting Canadians from Unsafe Drugs Act (Vanessa's Law), require hospitals to report serious adverse drug reactions to MHPD (see the Guidance Document: Mandatory reporting of serious adverse drug reactions and medical device incidents by hospitalsFootnote 22). These reports are also captured within this database.
If additional information is required (e.g. complete case narratives), the MAH is expected to request copies of AR reports through the Access to Information and Privacy Division of Health Canada (see appendix 4) and will require payment of the applicable fee. The MAH can also request line-listing summaries that were sent directly to Canada Vigilance Regional or National Offices (see Appendix 4 for contact information). Requests for line-listing summaries from the Canada Vigilance Adverse Reaction Database should be made in writing (letter, fax, or e-mail) to MHPD.
MAHs are not required to re-report cases associated with their marketed health products when identified from the Canada Vigilance database, unless there is new information relating to the causality assessment of a health product's safety and effectiveness. As such, MAHs are required to re-submit these cases only if new information (beyond what is already in the CV Online Database or in the copy obtained via the Access to Information Program), can be provided with the report to avoid duplicate reporting.
Reportable new information includes new additional information on key data elements such as patient gender, age or medical history, the batch/lot number or route of administration of the suspected health product, or additional descriptions of the reaction or reported signs and symptoms (see section 3.8 for a complete list of key data elements). The sole confirmation of the MAH's causality assessment for a case identified from the CV Online database would not be considered new information.
Reports that do not qualify for expedited regulatory reporting should however continue to be tracked by the manufacturer and captured in annual summary reports (ASRs), as highlighted in section C.01.018 of the Regulations.
As per the above, if the MAH has new information regarding a CV Online Database case meeting the criteria for expedited reporting, a new report must be submitted to MHPD within the prescribed reporting timelines, with the proper duplicate reference acknowledged.
4.3.1 Submitting an electronic report involving new information associated to a CV Online Database duplicate case
To aid in the identification of these duplicate reports when electronically resubmitting cases containing additional information, it is important to ensure that the guidance for duplicate reports from the CV Online Database be followed, such that the XML include for example:
<duplicate>1</duplicate>
<reportduplicate>
<duplicatesource>Canada Vigilance</duplicatesource>
<duplicatenumb>CA-HEALTHCANVIG-E2B_12345667</duplicatenumb>
</reportduplicate>
The duplicatenumb should be populated with the safetyreport ID of the duplicate report. The preferred format of the safety report ID is a concatenation of "country code-company or regulator name-report number".
4.3.2 Submitting a manual report involving new information associated to a CV Online Database duplicate case
For manual reporters, when submitting cases found on the Canada Vigilance Adverse Reaction Online Database containing additional information, please indicate on the report, the source of the report, including Canada Vigilance as the source, and quote the Canada Vigilance Adverse Event Report (AER) number, as well as any additional company case identification numbers of which the MAH is aware.
Lastly, when screening the CV Online Database, if potential duplicate reports are discovered, MAHs are strongly encouraged to notify the Canada Vigilance Program by email (see Appendix 4). The reports will be reviewed and may be linked as duplicates as necessary.
4.3.3 Courtesy Adverse Reaction Notifications
While conducting surveillance activities, MAHs may identify ARs to their marketed products as well as ARs to marketed products for which they do not hold the Drug Identification Number (DIN) or the Notice of Compliance (NOC). For example, a MAH may identify an individual case safety report in which there are multiple suspect products associated with an AR, of which the MAH only identifies one product as their own. Alternatively, a MAH identifies a literature report in which the brand name of the product is not identified and as such it could be the MAH's own product as well as the product of many other MAHs. In both cases, the MAH that retrieves the report is not required to exchange this information with other MAHs, as it is expected that each MAH will be conducting their own surveillance activities.
That being said, Health Canada acknowledges the value of sharing pharmacovigilance information between companies in the context of health product safety monitoring, but discourages this practice when a Safety Data Exchange Agreement is not in place between companies for the purpose of expedited reporting, as this may create false signals associated to multiple counting of a single event by various regulatory authorities, if not properly referenced.
As with any report submission to MHPD, we ask that you identify the source of the report accordingly, with any identifier number associated with the report, in such way to allow Health Canada to conciliate duplicate reports.
Appendix 1 Glossary: Definitions and Terminology
Adverse EventFootnote 8 (AE)
An adverse event is any untoward medical occurrence in a patient administered a medicinal product and which does not necessarily have to have a causal relationship with this treatment. An adverse event can therefore be any unfavourable and unintended sign (for example, an abnormal laboratory finding), symptom, or disease temporally associated with the use of a medicinal product, whether or not considered related to this medicinal product.
Adverse reaction (AR)
For the purpose of this guidance document means a noxious and unintended response to a marketed health product covered by this document and includes "adverse drug reaction" as defined in the Food and Drug Regulations and "adverse reaction" as defined in the Natural Health Products Regulations.
"Adverse Drug Reaction"
Adverse drug reaction as defined in the Food and Drug Regulations is a noxious and unintended response to a drug, which occurs at doses normally used or tested for the diagnosis, treatment or prevention of a disease or the modification of an organic function.
"Adverse Reaction"
Adverse reaction as defined in the Natural Health Products Regulations is a noxious and unintended response to a natural health product that occurs at any dose used or tested for the diagnosis, treatment or prevention of a disease or for modifying an organic function.
Brand Name (Food and Drug Regulations)
With reference to a drug, the name, whether or not including the name of any manufacturer, corporation, partnership or individual, in English or French,
- (a) that is assigned to the drug by its manufacturer,
- (b) under which the drug is sold or advertised, and
- (c) that is used to distinguish the drug.
Brand Name (Natural Health Products Regulations)
Means a name in English or French, whether or not it includes the name of a manufacturer, corporation, partnership or individual
- (a) that is used to distinguish the natural health product; and
- (b) under which a natural health product is sold or advertised.
Canada Vigilance Program
Health Canada's Canada Vigilance Program is responsible for the collection and assessment of adverse reaction reports related to the following marketed health products: pharmaceuticals, medical devices, natural health products, biologics (including biotechnology products, vaccines, DIN-assigned blood products, human blood and blood components, as well as cells, tissues and organs), radiopharmaceuticals, and disinfectants and sanitizers with disinfectant claims. The program is operated by the Marketed Health Products Directorate.
Common Name (Food and Drug Regulations)
With reference to a drug, the name in English or French by which the drug is
- (a) commonly known, and
- (b) designated in scientific or technical journals, other than the publications referred to in Schedule B to the Act.
Common Name (Natural Health Products)
For any medicinal or non-medicinal ingredient contained in a natural health product, the name by which it is commonly known and is designated in a scientific or technical reference.
Domestic AR
Adverse reaction occurring in Canada.
Drug
According to the Food and Drugs Act, a drug includes any substance or mixture of substances manufactured, sold or represented for use in:
- the diagnosis, treatment, mitigation or prevention of a disease, disorder or abnormal physical state, or its symptoms, in human beings or animals,
- restoring, correcting or modifying organic functions in human beings or animals, or
- disinfection in premises in which food is manufactured, prepared or kept.
Expected AR
An AR whose nature (i.e., specificity or outcome), severity or frequency is consistent with the terms or description used in the Canadian product labelling should be considered expected.
Expedited AR Report
The following must be reported by the MAH within 15 calendar days of receiving information:
- any serious domestic AR,
- any serious unexpected foreign AR, and
- any domestic unusual failure in efficacy for a new drug.
Foreign AR
An adverse reaction occurring outside Canada to a product with the same combination of active ingredients that is marketed in Canada irrespective of variations in the formulation, dosage form, strength, route of administration, or indication.
Health product
For the purpose of this guidance document, health products include the following products regulated under the Food and Drug Regulations ("drugs") and the Natural Health Products Regulations ("natural health products"):
- pharmaceutical drugs (which includes prescription and non-prescription pharmaceutical drugs);
- biologics as set out in Schedule D of the Food and Drugs Act (which include biotechnology products, vaccines and DIN-assigned manufactured blood products);
- radiopharmaceutical drugs listed in Schedule C of the Food and Drugs Act;
- natural health products as defined in Section 1 of the Natural Health Products Regulations.
Individual CaseFootnote 13
An Individual Case is an adverse reaction report, the information provided by a primary source to describe most completely suspected adverse reaction(s) to the use of one or more health products by an individual patient at a particular point in time.
Individual Case Safety Report (ICSR)Footnote 12
An ICSR is an Individual Case/adverse reaction report
Market authorization holder (MAH)
For the purpose of this guidance document means the entity that holds the Notice of Compliance, the Drug Identification Number (DIN), the Natural Product Number (NPN), the Homeopathic Medicine Number (DIN-HM), or the product licence.
Natural Health Product (NHP)
A substance set out in Schedule 1 of the Natural Health Products Regulations or a combination of substances in which all the medicinal ingredients are substances set out in Schedule 1 of the Natural Health Products Regulations, a homeopathic medicine or a traditional medicine, that is manufactured, sold or represented for use in
- the diagnosis, treatment, mitigation or prevention of a disease, disorder or abnormal physical state or its symptoms in humans;
- restoring or correcting organic functions in humans; or
- modifying organic functions in humans, such as modifying those functions in a manner that maintains or promotes health.
However, a natural health product does not include a substance set out in Schedule 2 of the Natural Health Products Regulations, any combination of substances that includes a substance set out in Schedule 2 of the Natural Health Products Regulations or a homeopathic medicine or a traditional medicine that is or includes a substance set out in Schedule 2 of the Natural Health Products Regulations.
New Drug
As per the Food and Drugs Regulations: Division 8 (C.08.001):
- a drug that contains or consists of a substance, whether as an active or inactive ingredient, carrier, coating, excipient, menstruum or other component, that has not been sold as a drug in Canada for sufficient time and in sufficient quantity to establish in Canada the safety and effectiveness of that substance for use as a drug;
- a drug that is a combination or two or more drugs, with or without other ingredients, and that has not been sold in that combination or in the proportion in which those drugs are combined in that drug, for sufficient time and sufficient quantity to establish in Canada the safety and effectiveness of that combination and proportion for use as a drug; or
- a drug, with respect to which the manufacturer prescribes, recommends, proposes or claims a use as a drug, or a condition of use as a drug, including dosage, route of administration, or duration of action and that has not been sold for that use or condition of use in Canada, for sufficient time and in sufficient quantity to establish in Canada the safety and effectiveness of that use or condition of use of that drug.
Product MonographFootnote 19 (PM)
A product monograph is a factual, scientific document on the drug product that, devoid of promotional material, describes the properties, claims, indications, and conditions of use for the drug, and that contains any other information that may be required for optimal, safe, and effective use of the drug.
Phase I StudyFootnote 20 (Drugs)
Clinical trials designed to determine the pharmacokinetics/pharmacological actions of the drug and the side effects associated with increasing doses. Drug interaction studies are usually considered as Phase I trials regardless of when they are conducted during drug development. Phase I trials are generally conducted in healthy volunteers, but may be conducted in patients when administration of the drug to healthy volunteers is not ethical.
Phase II StudyFootnote 20 (Drugs)
Clinical trials to evaluate the efficacy of the drug in patients with medical conditions to be treated, diagnosed or prevented, and to determine the side effects and risks associated with the drug. If a new indication for a marketed drug is to be investigated, then those clinical trials may generally be considered Phase II trials.
Phase III StudyFootnote 20 (Drugs)
Controlled or uncontrolled trials conducted after preliminary evidence suggesting efficacy of the drug has been demonstrated. These are intended to gather the additional information about the clinical efficacy and safety under the proposed conditions of use.
Phase IV StudyFootnote 20 (Drugs)
All studies performed after the drug has been approved by the regulator for the market, and related to the approved indication. These studies are often important for optimizing the drug's use. They may be of any type but must have valid scientific objectives. Commonly conducted studies include safety studies designed to support use under the approved indication such as mortality and morbidity studies, or epidemiological studies.
Phase IV StudyFootnote 21 (Natural Health Products)
All studies performed after the NHP has been approved by the regulator for the market and related to the approved conditions of use. These studies are often important for optimizing the NHP's use. They may be of any type but must have valid scientific objectives. Commonly conducted studies include safety studies and studies designed to support use under the approved conditions of use, such as mortality and morbidity studies or epidemiological studies.
Qualified Health Professional
A person who is a member in good standing of a professional medical, nursing, pharmacists' or other health care practitioner association and entitled to provide health care under the laws of the jurisdiction in which the person is located, or other individuals retained by the MAH who have the appropriate health care education and therapeutic expertise.
RegistryFootnote 13
An organized collection of data on humans within a particular disease, group or other special group (e.g., cancer, pregnancy, congenital anomaly or congenital disorder, organ transplant, and serious skin disease registries).
SellFootnote 18
Includes offer for sale, expose for sale, have in possession for sale and distribute, whether or not the distribution is made for consideration.
Serious Adverse Reaction
For the purpose of this guidance document means a noxious and unintended response to a marketed health product covered by this document that occurs at any dose and that requires in-patient hospitalization or prolongation of existing hospitalization, that causes congenital malformation, results in persistent or significant disability or incapacity, is life-threatening or results in death and includes "serious adverse drug reaction" as defined in the Food and Drug Regulations and "serious adverse reaction" as defined in the Natural Health Products Regulations.
"Serious Adverse Drug Reaction"
A serious adverse drug reaction as defined in the Food and Drug Regulations is a noxious and unintended response to a drug that occurs at any dose and that requires in-patient hospitalization or prolongation of existing hospitalization, causes congenital malformation, results in persistent or significant disability or incapacity, is life-threatening or results in death.
"Serious Adverse Reaction"
A serious adverse reaction as defined in the Natural Health Products Regulations is a noxious and unintended response to a natural health product that occurs at any dose and that requires in-patient hospitalization or a prolongation of existing hospitalization, that causes congenital malformation, that results in persistent or significant disability or incapacity, that is life threatening or that results in death.
It is important to note that, as per ICH standards, the above terms for a serious adverse drug reaction and serious adverse drug reaction could also refer to medically important events that may not be immediately life-threatening or result in death or hospitalization, but may jeopardize the patient or may require intervention to prevent one of the other serious outcomes above (e.g.: intensive treatment in an emergency room for allergic bronchospasm, blood dyscrasias or convulsions that do not result in hospitalization).
Serious Unexpected Adverse Drug Reaction (Food and Drug Regulations)
A serious adverse drug reaction that is not identified in nature, severity or frequency in the risk information set out on the label of the drug.
Serious Unexpected Adverse Reaction (Natural Health Products Regulations)
A serious adverse reaction that is not identified in nature, severity or frequency in the risk information set out on the label of the natural health product.
Solicited ReportFootnote 8
Solicited reports are those derived from organized data collection systems, which include clinical trials, registries, post-approval named patient use programs, other patient support and disease management programs, surveys of patients or health care providers, or information gathering on efficacy or patient compliance. Adverse event reports obtained from any of these should not be considered spontaneous.
Spontaneous Report
A spontaneous report is an unsolicited communication by a health professional or consumer to a company, regulatory authority or other organization (e.g., WHO, Regional Centre, Poison Control Centre) that describes one or more adverse reactions in a patient who was given one or more medicinal productsFootnote * and that does not derive from a study or any organized data collection scheme.
Stimulated Report
A report that may have been motivated, prompted or induced, such as advisories, literature report, publication in the press, or questioning of health professionals by MAH representatives. These reports should be considered unsolicited in nature.
Trading PartnerFootnote 12
An organization exchanging Electronic Data Interchange (EDI) messages in the area of pharmacovigilance in the pre- and post- authorization phase.
Unsolicited Report
See Spontaneous Report
- Footnote *
-
As extracted from ICH E2D. For the purposes of this guidance document, a medicinal product is a health product.
Appendix 2 References
- Footnote 1
-
Food and Drug Regulations, Part C, Division 1, C.R.C., c. 870.
- Footnote 2
-
Food and Drug Regulations, Part C, Division 8, C.R.C., c. 870.
- Footnote 3
-
Natural Health Products Regulations, Section 24, Reaction Reporting, C.R.C., SOR/2003-196.
- Footnote 4
-
Food and Drug Regulations, Part C, Division 5, C. R. C., c.870.
- Footnote 5
-
International Council on Harmonisation, Clinical Safety Data Management: Definitions and Standards for Expedited Reporting (ICH E2A) (1994).
- Footnote 6
-
International Council on Harmonisation, Maintenance of Clinical Safety Data Management including Data Elements for Transmission of Individual Case Safety Reports (ICH E2B(R2)) (2001).
- Footnote 7
-
International Council on Harmonisation, Periodic Benefit-Risk Evaluation Report (ICH E2C(R2)) (2012).
- Footnote 8
-
International Council on Harmonisation, Post-approval Safety Data Management: Definitions and Standards for Expedited Reporting (ICH E2D) (2003).
- Footnote 9
-
International Council on Harmonisation, Pharmacovigilance Planning (ICH E2E) (2004).
- Footnote 10
-
Council for International Organizations of Medical Sciences, Current Challenges in Pharmacovigilance: Pragmatic Approaches, Report of CIOMS Working Group V (Geneva, Switzerland: CIOMS, 2001), 72.
- Footnote 11
-
Natural Health Products Regulations, Interpretation, C.R.C., SOR/2003-196.
- Footnote 12
-
Health Canada, Instructions for Market Authorization Holders & Clinical Trial Sponsors on Electronic Reporting of Adverse Reactions to Canada Vigilance (2014).
- Footnote 13
-
Council for International Organizations of Medical Sciences.
- Footnote 14
- Footnote 15
-
Health Canada, Good Pharmacovigilance Practices (GVP) Guidelines GUI-0102 (2013).
- Footnote 16
-
International Council on Harmonisation, MedDRA®Term Selection: Points to Consider.
- Footnote 17
-
International Council on Harmonisation, MedDRA®Data Retrieval and Presentation: Points to Consider.
- Footnote 18
-
Food and Drugs Act, Revised Statutes of Canada, Interpretation, 1985, c. F-27, as amended.
- Footnote 19
- Footnote 20
-
Health Canada, Guidance for Clinical Trial Sponsors: Clinical Trial Applications (2013).
- Footnote 21
-
Health Canada, Clinical Trials for Natural Health Products (NHPs) (2005).
- Footnote 22
- Footnote 23
- Footnote 24
- Footnote 25
- Footnote 26
-
Health Canada: Guidance Document for Mandatory Problem Reporting for Medical Devices (2011).
Appendix 3 Abbreviations
- AE
- Adverse Event
- AER
- Adverse Reaction Report Number
- AR
- Adverse Reaction
- ASR
- Annual Summary Report
- ATI
- Access to Information
- BBRDD
- Biologics and Radiopharmaceutical Drugs Directorate
- CAMR
- Canada's Access to Medicines Regime
- CIOMS
- Council for International Organizations of Medical Sciences
- CTA
- Clinical Trial Application
- CTO
- Cells, Tissues, and Organs
- DIN
- Drug Identification Number
- DIN-HM
- Homeopathic Medicinal Number
- EDI
- Electronic Data Interchange
- FDR
- Food and Drugs Regulations
- GVP
- Good Pharmacovigilance Practices
- HC
- Health Canada
- HPC
- Health Professional Communication
- HPFB
- Health Products and Food Branch
- ICH
- International Council on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use
- ICSR
- Individual Case Safety Report
- INN
- International Nonproprietary Name
- MAH
- Market Authorization Holder
- MedDRA
- Medical Dictionary for Regulatory Activities
- MHPD
- Marketed Health Products Directorate
- NHP
- Natural Health Product
- NNHPD
- Natural and Non-prescription Health Products Directorate
- NHPR
- Natural Health Products Regulations
- PC
- Public Communication
- PM
- Product Monograph
- TPD
- Therapeutic Products Directorate
- TPMO
- Trading Partner Management Office
- The Regulations
- Collectively, the Food and Drug Regulations and the Natural Health Products Regulations
- WHO
- World Health Organization
Appendix 4 Contact Information
Electronic Reporting – Trading Partner Management Office (TPMO)
The preferred method of reporting ARs is electronically. The TPMO provides a single point of contact for trading partners already enrolled with Canada Vigilance Program to submit ARs electronically as well as for MAHs seeking enrolment. For the guidance document on e-reporting, contact TPMO.
During normal business hours, the TPMO may be reached at:
HPFB Trading Partner Management Office
Health Canada
250 Lanark Avenue
Address Locator 2004D
Ottawa, Ontario
K1A 0K9
E-mail: hc.tpmo_bgpc.sc@canada.ca
Non-Electronic Reporting
For MAHs who are not yet enrolled as a trading partner, AR reports for marketed health products covered by this guidance document should be sent to:
Canada Vigilance Program
Health Canada
Address Locator 1908C
200 Eglantine Driveway
Ottawa, Ontario
K1A 0K9
Telephone: 613-957-0337
Facsimile: 613-957-0335
E-mail: hc.canada.vigilance.sc@canada.ca (DO NOT SEND REPORTS VIA E-MAIL)
Access to Information
For copies of AR reports, consult the Access to Information website at:
Access to Information and Privacy
Council for International Organizations of Medical Sciences (CIOMS)
CIOMS publications may be obtained directly from:
Council for International Organizations of Medical Sciences (CIOMS)
Case postale 2100
CH-1211 Geneva 2
Switzerland
Telephone: + 41 22 791 6497
Email: info@cioms.ch
Website: www.cioms.ch
International Council on Harmonisation (ICH)
ICH guidance documents may be obtained from:
ICH Secretariat
c/o IFPMA
9, chemin des Mines
1202 Geneva
Switzerland
Telephone: +41 22 338 3207
Email: admin@ich.org
Website: http://www.ich.org
Appendix 5 Other Adverse Reaction Reporting Programs Outside the Scope of this Document
Canada Vigilance Program and its partners collect adverse reaction reports in order to monitor health and safety risks related to the sale and use of a variety of products. In order to avoid delays in reporting, it is important to direct adverse reaction reports to the appropriate program area of expertise.
Refer to the following link which provides further information on adverse reaction reporting specific to other products that are not within the scope of this guidance document. All adverse reaction reports related to marketed health products should be sent to the Canada Vigilance national office (see Appendix 4).
Adverse Reaction Reporting for Specific Products
Appendix 6 World Health Organization Causality Algorithm
Refer to the following link for additional information on the WHO and the Centre for International Drug Monitoring (Uppsala Monitoring Centre).
| Term | Description | Comments |
|---|---|---|
| Certain | A clinical event, including laboratory test abnormality, occurring in a plausible time relationship to drug administration, and which cannot be explained by concurrent disease or other drugs or chemicals. The response to withdrawal of the drug (dechallenge) should be clinically plausible. The event must be definitive pharmacologically or phenomenologically, using a satisfactory rechallenge procedure if necessary. | It is recognized that this stringent definition will lead to very few reports meeting the criteria, but this is useful because of the special value of such reports. It is considered that time relationships between drug administration and the onset and course of the adverse event are important in causality analysis. So also is the consideration of confounding features, but due weight must placed on the known pharmacological and other characteristics of the drug product being considered. Sometimes the clinical phenomena described will also be sufficiently specific to allow a confident causality assessment in the absence of confounding features and with appropriate time relationships, e.g. penicillin anaphylaxis. |
| Probable / Likely | A clinical event, including laboratory test abnormality, with a reasonable time sequence to administration of the drug, unlikely to be attributed to concurrent disease or other drugs or chemicals, and which follows a clinically reasonable response on withdrawal (dechallenge). Rechallenge information is not required to fulfil this definition. | This definition has less stringent wording than for "certain" and does not necessitate prior knowledge of drug characteristics or clinical adverse reaction phenomena. As stated no rechallenge information is needed, but confounding drug administration underlying disease must be absent. |
| Possible | A clinical event, including laboratory test abnormality, with a reasonable time sequence to administration of the drug, but which could also be explained by concurrent disease or other drugs or chemicals. Information on drug withdrawal may be lacking or unclear. | This is the definition to be used when drug causality is one of other possible causes for the described clinical event. |
| Unlikely | A clinical event, including laboratory test abnormality, with a temporal relationship to drug administration which makes a causal relationship improbable, and in which other drugs, chemicals or underlying disease provide plausible explanations. | This definition is intended to be used when the exclusion of drug causality of a clinical event seems most plausible. |
| Conditional / Unclassified | A clinical event, including laboratory test abnormality, reported as an adverse reaction, about which more data is essential for a proper assessment or the additional data are under examination. | N/A |
| Unassessable / Unclassifiable | A report suggesting an adverse reaction which cannot be judged because information is insufficient or contradictory, and which cannot be supplemented or verified. | N/A |
Appendix 7 Summary of Expedited Post-Market AR Reporting Requirements to MHPD
| Reports | Types of Reaction | Drug and Natural Health Product ARs to be Reported to MHPD Within 15 Calendar Days |
|---|---|---|
| Unsolicited Domestic Reports | Serious unexpected | Yes |
| Serious expected | Yes | |
| Non-serious unexpected | No | |
| Non–serious expected | No | |
| Solicited Domestic Reports (e.g., StudiesFootnote *) | Serious unexpected | Yes |
| Serious expected | Yes | |
| Non-serious unexpected | No | |
| Non–serious expected | No | |
| Unusual failure in efficacy for new drugs | Yes | |
| Unsolicited Foreign Reports | Serious unexpected | Yes |
| Serious expected | No | |
| Non-serious unexpected | No | |
| Non–serious expected | No | |
| Solicited Foreign reports (e.g., StudiesFootnote *) | Serious unexpected | Yes |
| Serious expected | No | |
| Non-serious unexpected | No | |
| Non–serious expected | No | |
| Unusual failure in efficacy for new drugs | No | |
|
||
Appendix 8 Determining when to report cases found in the Canada Vigilance Adverse Reaction Online Database
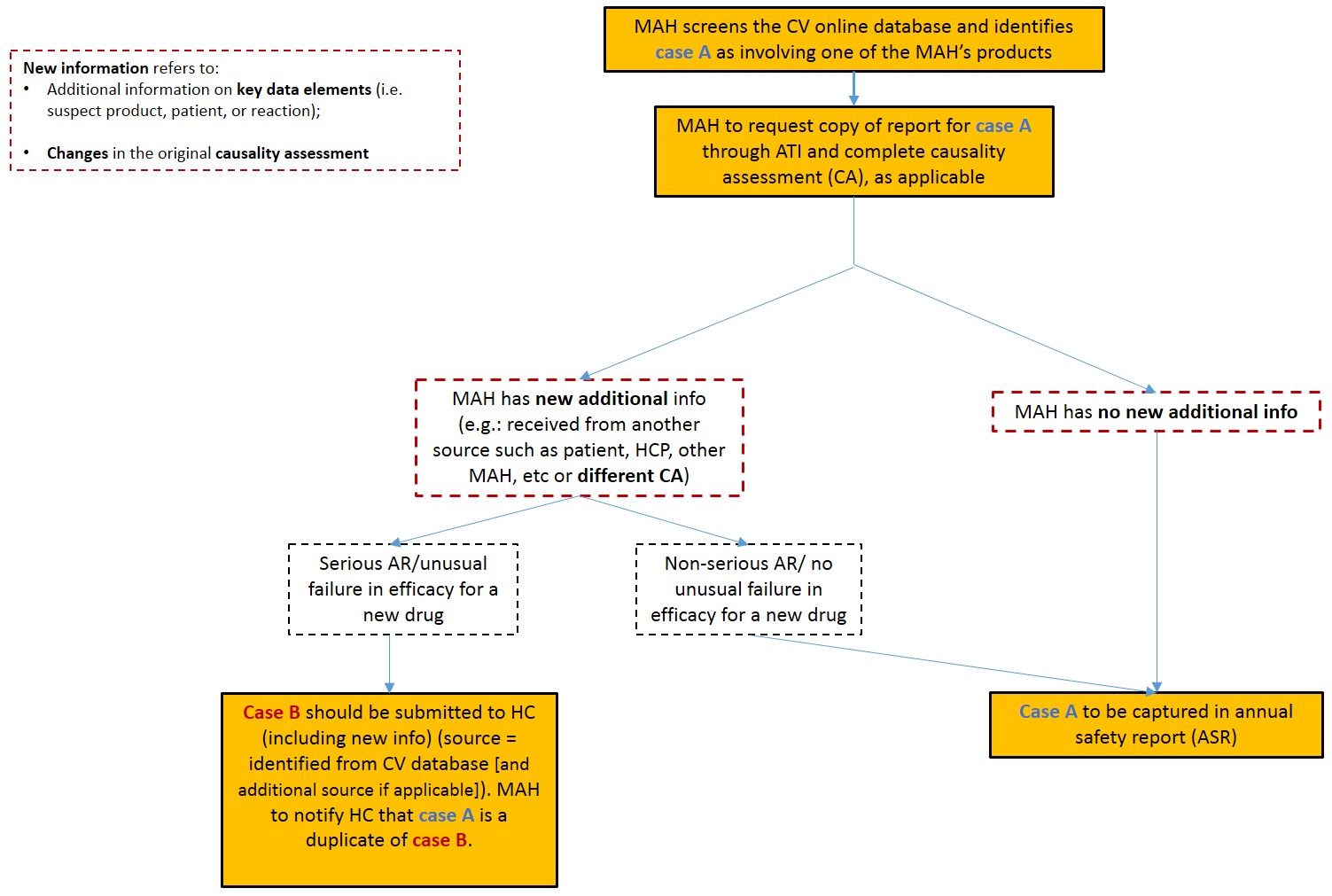
Determining when to report cases found in the Canada Vigilance Adverse Reaction Online Database - Text description
If a MAH screens the Canada Vigilance (CV) Adverse Reaction Online Database and identifies case A as involving one of the MAH's products, the MAH is encouraged to request a copy of the report for case A through the Access to Information (ATI) Program and complete a causality assessment (CA), as applicable. Thereafter, if the MAH determines they have no new additional information, case A should be captured in the annual safety report (ASR) and expedited reporting is not required. Alternatively, if the MAH determines they have new additional information (e.g. information received from another source such as a patient, health care professional (HCP), hospital, other MAH, or a different CA), with respect to a serious AR or an unusual failure in efficacy for a new drug, case B should be submitted to Health Canada (HC) in an expedited fashion. Case B should include the new information, the source should be identified as the CV Online Database, as well as additional sources if applicable, and the MAH is to notify HC that case A is a duplicate of case B. However, if the MAH has new additional information with respect to an AR case that is non-serious and that does not refer to an unusual failure in efficacy for a new drug, case B is to be captured in the ASR and submitted to HC.
Page details
- Date modified: