
Growth and tolerance clinical trial protocol – preterm infants
A guidance document for preterm infant formula and human milk fortifier manufacturers
2021
Acknowledgement: Thank you to Dr. Tanis Fenton’s contribution in peer-reviewing this guidance document.
Table of contents
- 1.0 Introduction
- 2.0 Definitions
- 3.0 Protocol for growth and tolerance clinical trials to support preterm infant formula/human milk fortifier premarket submissions
- 3.1 Registration
- 3.2 Study title
- 3.3 Abstract
- 3.4 Introduction and background
- 3.5 Study design
- 3.5.1 Description of study design
- 3.5.2 Study objectives
- 3.5.3 Choice of non-inferiority/superiority margin
- 3.5.4 Subject eligibility
- 3.5.5 Sample size
- 3.5.6 Intervention
- 3.5.7 Study duration
- 3.5.8 Assignment
- 3.5.9 Blinding
- 3.5.10 Data collection and outcomes
- 3.5.11 Adverse events
- 3.5.12 Stopping rules, discontinuation, withdrawals and dropouts
- 3.5.13 Quality control procedures
- 3.5.14 Independent Data Monitoring Committee (IDMC)
- 3.5.15 Protocol violation
- 3.5.16 Protocol amendment
- 3.5.17 Pre-planned statistical analyses
- 3.6 Study results
- 3.7 Discussion and conclusions
- 4.0 Ethical standard for investigators
- 5.0 Glossary and references
- Appendix 1. CONSORT 2010 checklist of information to include when reporting a randomized trial
- Appendix 2. Biochemical markers of protein and bone status in preterm infants
- Appendix 3. Nutrient requirement template for preterm infant formula or fortifier with human milk (as fed) per 100 kcal
- Appendix 4. Summary of fluids and energy intakes recommended by international expert committees
- Appendix 5. Preterm human breast milk composition
1.0 Introduction
All preterm infant formulas and human milk fortifiers must be safe and fit for human consumption as per Section 4.1 of the Food and Drugs Act. These foods, intended for the infant population, are regulated as per the Food and Drug Regulations (FDR), Part B Division 25, and require a premarket assessment by Health Canada.
Petitioners must submit a premarket notification for a new preterm infant formula or human milk fortifier, or one that has undergone a major change in composition, manufacturing, or packaging. Notifications must include the evidence relied upon to establish that the preterm infant formula or human milk fortifier is safe, nutritionally adequate and will promote acceptable growth and development in preterm infants when consumed in accordance with the directions for use.
The objective of this document is to provide guidance to manufacturers on the appropriate design of a clinical study for growth and tolerance. Health Canada encourages manufacturers to consult with Health Canada on the clinical protocol for any planned clinical studies, and to schedule a pre-submission meeting to review the evidence required for premarket assessment, since requirements may vary.
2.0 Definitions
- Chronological age
The age of the infant in weeks from the date of birth without correcting for prematurity (refer to Figure 1).
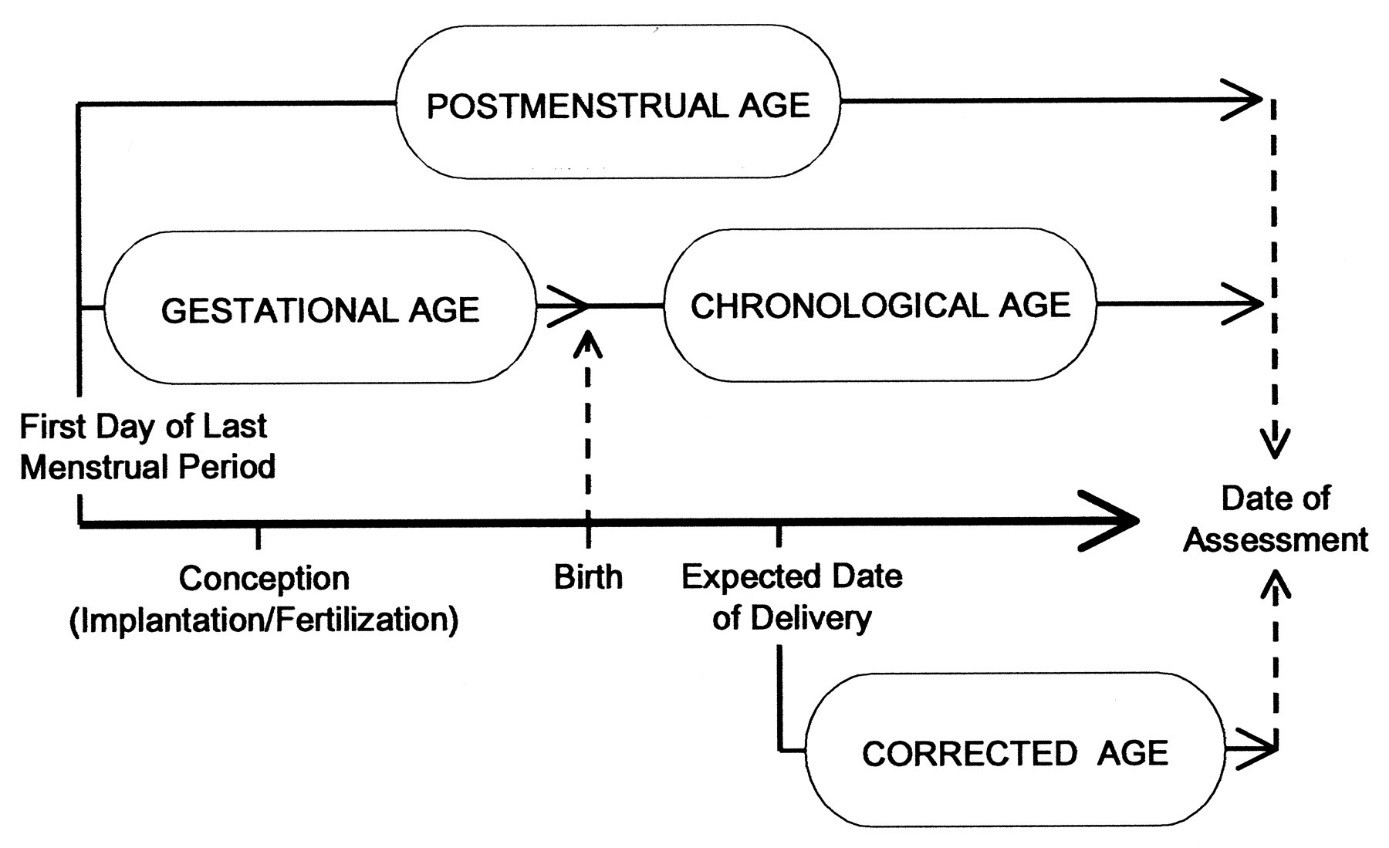
Figure 1 - Text description
A timeline showing the relationship between Postmenstrual age, Gestational age, chronological age and Corrected age, as defined in Section 2.0
- Control fortified human milk
Means an approved fortifier in Canada plus preterm human milk.
Note: Human milk fortifier is never clinically assessed alone. The combination of a fortifier + human milk is the final product tested. In this document, we refer to the combination of both the fortifier and the human milk as fortified human milk.
- Corrected age [week or months] (i.e. corrected for prematurity)
Means the chronological age reduced by the number of weeks born before 40 weeks of gestation; the term should be used only for children up to 3 years of age who were born preterm (1) (refer to Figure 1).
- Enteral feeding
Administration of any feed into the gastrointestinal tract; it includes intragastric feeding and cup, bottle and breastfeeding (2).
- Fortified human milk
Means human milk fortifier + preterm human milk.
- Gestational age
In obstetrics, the age of a fetus, beginning on the presumed first day of the last normal menstrual period (refer to Figure 1) (3).
- Human Milk Fortifier (FDR B.25.001)
Means a food that:
- includes at least one added vitamin, mineral nutrient or amino acid, and
- is labelled or advertised as intended to be added to human milk to increase its nutritional value in order to meet the particular requirements of an infant in whom a physical or physiological condition exists as a result of a disease, disorder or abnormal physical state.
- Intrauterine growth restriction (IUGR)
Failure to sustain intrauterine growth at expected rates; can be caused by placental insufficiency, infection, malnutrition, etc. May or may not be born prematurely. It describes a fetus that has not reached its growth potential because of genetic or environmental factors (4).
- Major Change for a human milk substitute, (FDR B.25.001)
Means, in respect of a human milk fortifier or a human milk substitute, any change of an ingredient, the amount of an ingredient or the processing or packaging of the human milk fortifier or human milk substitute where the manufacturer's experience or generally accepted theory would predict an adverse effect on the levels or availability of nutrients in, or the microbiological or chemical safety of, the human milk fortifier or human milk substitute.
Examples of major changes for a preterm infant formula or human milk fortifier:
- substantial change in processing or packaging of the preterm infant formula or HMF change of or a new manufacturing facility
- addition of a new macronutrient source (protein, fat, or carbohydrate)
- substantial change in the amount of protein, fat, or carbohydrate
- addition of a new ingredient or novel food
- change in the amount or the source of vitamins or minerals, which, in the manufacturer's experience or generally accepted theory, would predict an adverse effect on:
- the levels or availability of the nutrients, or
- the microbiological or chemical safety
For more information about the scientific evidence needed to establish the nutritional adequacy of a new preterm infant formula or human milk fortifier, or a preterm infant formula or human milk fortifier that has undergone a major change, please refer to the guide, Scientific Evidence Requirements for Nutritional Adequacy- Preterm Infant Formulas and Human Milk Fortifiers.
- New human milk substitute (Infant Formula) (FDR B.25.001)
Means a human milk substitute that is:
- manufactured for the first time, or
- sold in Canada for the first time, or
- manufactured by a person who manufactures it for the first time.
- Oral feeding
Administration of any feed into the oral cavity; it includes cup, spoon, syringe, direct expression, bottle and breastfeeding but not gastric tube feeding (2).
- Parenteral nutrition
Often called total parenteral nutrition, is the medical term for infusing a specialized form of food through a vein (intravenously). The goal of the treatment is to correct or prevent malnutrition.
- Postmenstrual age (PMA)
Indicates the time elapsed between the first day of the last menstrual period and birth (gestational age) plus the time elapsed after birth (chronological age) in weeks and days. This is the preferred term used to describe the age of the preterm infant during the perinatal period and during hospital stay. After the perinatal period, "corrected age" is the preferred term (1)(refer to Figure 1).
- Postnatal growth failure
Is a decrease in weight between birth and discharge of more than −2 z-scores using the Fenton Preterm Growth Chart (5).
- Prematurity
Refers to the broad category of neonates born at a gestational age of less than 37 weeks (3). The majority of preterm births fall between 34-36 weeks and account for 60-70% of all preterm births, 15% of preterm births fall between 28-31 weeks, and 5% at less than 28 weeks (6). Preterm infants can also be classified further according to their birth weight, or their weight for gestational age as outlined in Table 1. In addition, every category is further subdivided into different functional maturation stages postnatal: transition, stabilization and established growth.
Premature and low birth weight (LBW) infants have many physiological challenges when adapting to the extrauterine environment and are generally admitted at birth to a Neonatal Intensive Care Unit (NICU) in order to stabilize their conditions and to allow for specialized medical treatment and nutritional care. The purpose of care in the NICU is to assess and monitor the needs of each preterm or LBW infant individually and to provide appropriate support until functional maturity can be achieved (7).
- Preterm infant
Is defined as a baby born alive before 37 weeks of pregnancy are completed. There are sub-categories of preterm birth, based on gestational age (3)(refer to Table 1).
| Classification scheme | Group | Definition |
|---|---|---|
| Gestational age(3) | Extreme prematurity/micropreterm | < 28 weeks |
| Severe prematurity | 28 - < 32 weeks | |
| Moderate to late prematurity | 32 - < 37 weeks | |
| Birth weight(8) | Low birth weight (LBW) | < 2500 g |
| Very low birth weight (VLBW) | < 1500 g | |
| Extremely low birth weight (ELBW) | < 1000 g | |
| Weight for age(8) | Small for gestational age (SGA) | Birth weight < 10th percentile of used preterm growth charts reference |
| Appropriate for gestational age (AGA) | Birth weight ≥ 10th and ≤ 90th percentile of used preterm growth charts reference | |
| Large for gestational age (LGA) | Birth weight > 90th percentile of used preterm growth charts reference |
- Preterm infant formula
Means any food that is represented for use as a partial or total replacement for preterm human milk and intended for consumption by preterm, LBW, VLBW and ELBW infants. Preterm infant formulas are enriched in calories (approximately 80 kcal/100ml vs. 67 kcal/100 ml for term formulas), protein and minerals to help preterm infants approximate intra-uterine nutrient accretion rates. The calories are primarily provided as protein, fat and carbohydrate. The latter two macronutrients are used to increase energy density. The balance between calories and protein is important to support the growth of lean tissues mass. Compared to unfortified human milk or standard infant formula, preterm infant formulas contain more protein, sodium, calcium, phosphorus, zinc, copper and vitamins.
- Recommended rate of weight gain
A goal of 15 - 20 g/kg/day calculated after the postnatal weight loss phase, most often around 7 days after birth, using either the exponential model or 2-point model for the average weight gain calculation (9,10,11, 12). This rate may be suitable for infants who are between 23 - 36 weeks postmenstrual age (12, 13).
- Stable growing period
The period beginning when the preterm infant is metabolically and clinically stable and growing, ending when the infant reaches 37 weeks of postmenstrual age (2) or hospital discharge.
- Test fortified human milk
Means the new or changed fortifier + preterm human milk.
- Transition period
The period from birth to 7 days when preterm infants are likely to be clinically and metabolically unstable and to lose weight (2).
3.0 Protocol for growth and tolerance clinical trials to support preterm infant formula/human milk fortifier premarket submissions
The following sections discuss all the general requirements for a clinical trial protocol for a growth and tolerance study in clinically and metabolically stable preterm, LBW or VLBW infants. These three categories of preterm infants represent the study population that will be referred to in the protocol as preterm infants.
The study report should follow the format specified in Appendix 1 (CONSORT 2010 checklist) (14).
3.1 Registration
Registration of clinical trials should meet the following requirements:
- The protocol should be registered on a clinical trial registry. Examples of acceptable trial primary registries are available on the WHO website (15);
- The registry should include all information and criteria specified in the World Health Organization Trial Registration Data Set (16);
- Registration number and name of trial registry should be reported;
- Registration should indicate where the full trial protocol can be accessed (17);
- Although changes in protocol after commencement are greatly discouraged, the clinical trial's registration must be updated when amendments or protocol violations are made after the trial commencement and details should be clearly described in the trial report (18).
3.2 Study title
The study title should specify that the study is a randomized, non-inferiority trial in preterm infants (18, 19).
A superiority trial is appropriate for assessment of a new preterm infant formula or human milk fortifier proposed to improve growth, feeding tolerance, or other health outcome, compared with the concurrent control.
3.3 Abstract
The protocol and study report should include an abstract that describes the trial objectives, design and methodology, data collection and analysis, and study findings/conclusions (14).
3.4 Introduction and background
The introduction should include the following information:
- name and description of the investigational product(s), including the physical form (e.g., example powder, concentrated liquid) and confirmation that the concurrent control is appropriate;
- rationale for the clinical trial and a clearly stated study question;
- description of the preterm population to be studied and its similarity with the target population;
- a summary of findings from relevant clinical and non-clinical studies that relate to preterm infants;
- a summary of known and potential risk and benefits, if any, to human subjects, particularly to preterm infants.
3.5 Study design
3.5.1 Description of study design
The study should be designed based on the gold standard for clinical trials: a prospective, randomized, well-controlled, double-blind, parallel and single/multi-center study (20). The inclusion of a schematic diagram of the trial design, procedures, and stages is required (14).
3.5.2 Study objectives
The objectives of the study should be to assess the effects of the test preterm infant formula (T) or fortified human milk on measures of growth, feeding tolerance, and safety. It should be done in comparison to an appropriate concurrent control (C) that has previously been clinically tested and has demonstrated safety and nutritional adequacy according to the acceptable Growth Charts for Preterm Infant (21).
3.5.2.1 Primary objective
The primary objective of the trial should be to assess the nutritional adequacyFootnote 1 as measured by weight gain rate over a minimum period of 28 days or until hospital discharge (beginning at 7 days after birth or when on full feeds of the product). This should be expressed as g/kg/day [calculated using either the exponential model or 2-point model for the average weight gain calculation (9,10,11,21,22) in preterm infants fed with the test preterm formula or fortified human milk as compared to those fed an appropriate concurrent control (23).
The nutritional goal for preterm infants is to achieve rates of growth and nutrient accretion that match those achieved by fetuses of similar postmenstrual age in utero, while maintaining normal concentrations of nutrients in blood and other tissues (24) and avoiding complications that can be caused by aggressive nutritional therapies.
Footnotes
- Footnote 1
-
For more information on the evidence required by Health Canada to support the nutritional adequacy of preterm infant formula or human milk fortifier, please refer to Scientific evidence requirements for nutritional adequacy of a preterm infant formula and human milk fortifiers – A guidance document for infant formula manufacturers
3.5.2.2 Secondary objectives
The secondary objectives should be to assess and compare the following growth and safety measures between the test and control groups:
- time to regain birth weight;
- rates of gain in length, and in head circumference, expressed as cm/week;
- attained weight, length and head circumference measurements recorded at all pre-specified intervals (weekly) during the study period, of individual and of groups, stratified by sex, birth weight and/or gestational age category and plotted on the Fenton Preterm Growth Charts for that growth measurement and sex category (21);
- group's standardized z-scores and changes in z-scores (and their 95% confidence intervals) for weight, length, and head circumference-for-age and weight-for length based on Fenton Preterm Growth Charts (12,21) at birth, study baseline and at each study visit by sex, birth weight and/or gestational age category;
- time to full enteral feeding defined as 150 ml/kg/day (25);
- time to reach 1800 - 2000 g body weight which is considered an important milestone in this area of research (potential time for hospital discharge);
- average daily intake volume of preterm infant formula or fortified human milk (mother's own milk or donor milk), calories, protein, modular supplements, vitamins and mineral supplementation;
- incidence of weight loss of more than 2 z-scores relative to the Fenton Preterm Growth Charts (21) at 36 weeks postmenstrual age or at discharge (5);
- duration of total parenteral nutrition (TPN) (days);
- duration of hospital stay (days);
- proportion of infants with at least 1 episode of feed interruption lasting ≥ 12 hours;
- physicians' blinded appraisal of tolerance, e.g., withhold of enteral feeding, gastric residuals, abdominal distention, colic, vomiting, fussiness, cramps, sleeping patterns, regurgitation and stool characteristics;
- incidence of adverse events and serious adverse events such as mortality, metabolic acidosis, renal failure/insufficiency, deteriorating respiratory status, sepsis, retinopathy of prematurity and necrotising enterocolitis (NEC), etc. recorded on a case report form (CRF) by health care professionals;
- any planned biochemical tests at baseline, mid point and at the end of the study such as serum albumin, blood urea nitrogen (BUN), creatinine, calcium, phosphorus, magnesium, hemoglobin, hematocrit, serum iron, total iron binding capacity, serum alkaline phosphatase, serum electrolytes, blood gases and pH, etc. The need for such tests depends on the nature of the formulation and would be determined on a case-by-case basis (refer to Appendix 2).
3.5.3 Choice of non-inferiority/superiority margin
At least one adequately powered clinical study in the target population should be conducted to assess the safety and nutritional adequacy of the preterm infant formula (T) or human milk fortifier. The design of the clinical trial depends on many factors and the study purposes.
A non-inferiority trial, with a predefined margin of non-inferiority (MNI), is highly recommended when the objective of the study is to assess safety and nutritional adequacy in preterm infants fed the test formula or fortified human milk as compared to those fed the commercial control formula or human milk fortifier (19, 20).
A superiority design with a predefined superiority margin (26) is recommended, if the aim of the study is to demonstrate the superiority of a new improved test formula or fortified human milk compared to an approved control.
In the case that a non-inferiority trial is originally conducted and the results of the trial show sufficient evidence of superiority, it is acceptable to switch the objective from non-inferiority to superiority. This could be achieved by calculating and evaluating the p-value associated with a test of superiority in the intention-to-treat (ITT) analysis (refer to "Switching from a non-inferiority trial to a superiority trial" section below).
Non-inferiority trial
A non-inferiority trial aims to show that a new test formula or fortified human milk is not inferior to the control preterm formula or fortified human milk. The test hypothesis concerning non-inferiority of weight gain rate in infants fed the test formula or fortified human milk compared to those fed the control should be clearly stated, specifying the MNI, which should be clinically justified (19).
It is very important to define a priori the MNI. The determination of the MNI is based on a combination of both statistical considerations and clinical judgement. The margin chosen for a non-inferiority trial must not be greater than the smallest effect size of the control product. Magnitude and variability of the effect size of the control should be derived from historical trials and should be taken into consideration [26]. Furthermore, the selected MNI should reflect uncertainties in the evidence on which the choice is based, be suitably conservative and account for variability as well (19, 26). Rationale and/or scientific literature to justify the choice of such margin should be included in the protocol.
In the past decade, several clinical trials of growth and tolerance in preterm infants have selected a mean difference that varied from -1 to -4 g/kg/day as a clinically acceptable MNI in weight gain rate (denoted as -δ) (27,28,29). Overall, a clinically acceptable MNI of -1.6 g/kg/day has been used more often than other values (29, 30, 31, 32).These clinical studies, however, did not sufficiently provide historical evidence or data to support their choices of the MNI in the weight gain rate. Recent systematic reviews and meta-analyses using the pooled data from several studies of growth and tolerance in preterm infants show that the MNI of -1.1 g/kg/day would be more reasonable and acceptable since it is based on clinical data (33,34,35). This MNI is within the range of half of the mean difference in weight gain rate and its lower bound (33,34), as recommended by the Food and Drug Administration (26). It is also equivalent to the recommended 0.25 standard deviation of weight gain rate (36), where the standard deviation of weight gain rate in preterm infants over a 4-week study period, starting between 7 and 21 days of life, is about 4.2 g/kg/day (36). The standard deviation of 4.2 g/kg/day, found in the recent systematic reviews and meta-analyses, is comparable to those reported (12,27,28). The hypothesis of the test product (T) being non-inferior to the control product (C) could be established if the lower bound of the 95% CI for the difference in weight gain rate between the 2 study arms (T- C) lies above the MNI of -1.1 g/kg/day as shown in Figure 2 (36,37).
Mean difference in weight gain rate (T-C, expressed in g/kg/day)
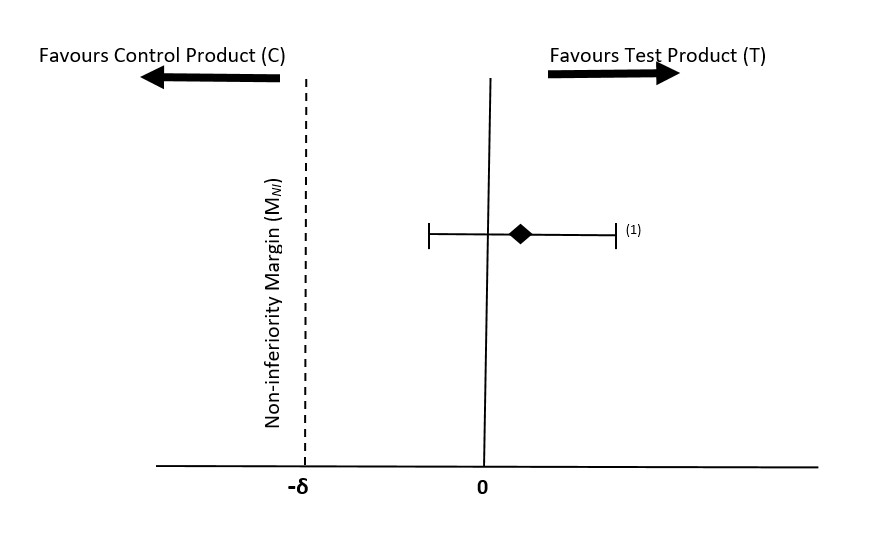
Figure 2 - Text description
Illustrates a possible outcome of a non-inferiority study, showing that the test product is non-inferior to the control product.
Superiority trial
A superiority trial aims to demonstrate the superiority of a new test formula or fortified human milk to an approved control formula or fortified human milk. The test hypothesis concerning superiority of growth rates in infants fed the test formula or fortified human milk compared to those fed the control(s) should be clearly stated, specifying the minimal clinically important difference in growth between infants fed the test formula or fortified human milk and those fed the controls, δ, and how large the risks of type I (α) and type II (β) errors would be acceptable for the trial. To achieve superiority, several outcomes must be assessed with no evidence of any adverse impacts on any important clinical primary or secondary endpoints.
Often a non-significant superiority test is wrongly interpreted as proof of no difference between the two treatments. In fact, the non-significant superiority test could be a result of not having enough statistical power (e.g., 80% power) to detect the differences between the test and control products. Therefore, it is very important to predefine the values used for α, β and δ in order to detect the true difference if it exists. Clinicians, researchers and statisticians should decide these values a priori. Most often, α, and β would be specified as 5% and between 10% and 20%, respectively, but in special situations, higher or lower values for type I and II errors may be used. The value of δ should be decided based on clinical grounds a priori as the least relevant of weight gain rate from the use of new test formula or fortified human milk, as well as the efficacy of the controls and what may reasonably be expected of additional effect from the new test formula or fortified human milk. Preliminary data from pilot studies or historical observational data can be used as a guidance for the choice of δ. Rationale and/or scientific literature to justify the choice of such values of α, β, and δ should be included in the protocol.
At a minimum, the same margin (MNI) of 1.1 g/kg/day and weight gain rate (δ) of 4.2 g/kg/day mentioned in the non-inferiority trial (see section above) should be applied for superiority trial.
Switching from a non-inferiority trial to a superiority trial
If the outcomes from an ITT analysis of a non-inferiority trial show that the lower limit of the 95% confidence interval for the treatment effect not only lies entirely above -δ (Figure 2) but also above zero, this constitutes potential evidence of superiority. In this case the p-value and power calculation associated with a test of superiority must be evaluated to determine whether there is sufficient evidence to reject the hypothesis of no difference in favor of superiority, provided that the safety profiles of the test and control formulas or fortified human milk are similar.
Overall, switching the objective of a trial from non-inferiority to superiority is feasible if the following requirements are fulfilled:
- the trial has been properly designed and carried out in accordance with the strict requirements of a non-inferiority trial and follows the CONSORT guidelines;
- actual p values for superiority are presented to allow independent assessment of the strength of the evidence;
- analysis according to the ITT principle is given greatest emphasis in superiority trial (38);
- there are no clinical or statistical increase in important adverse events.
3.5.4 Subject eligibility
Study subjects should be preterm infants, LBW or VLBW, with <37 weeks’ gestational age and hospitalized in NICUs. For ethical reasons, for preterm infant formula clinical trial, the mothers must have already decided not to breastfeed their infants. Methods of recruitment, clinical settings and geographical locations where the eligibility criteria are to be collected should be reported (39). Dates defining the periods of recruitment and follow-up, information on how these subjects were intended to be followed-up should be provided. Both male and female infants should be equally included in the study in numbers that conform to the power calculation.
The following inclusion and exclusion criteria should be applied:
Inclusion criteria
- clinically and metabolically stable preterm/LBW or VLBW;
- single/twin or triplet infants of <37 weeks gestation (both twins or all triplet do not need to qualify and be randomized into study);
- birth weight: ≤1800 g;
- appropriate for gestational age (see table above for definition);
- expected to be hospitalized for a minimum of the study duration after study enrollment;
- maternal intent to provide breast milk during the study in case of testing a new fortified human milk (allow donor milk, as needed);
- less than or equal to 21 days of age at enrolment;
- predominant formula or fortified human milk feeding before initiation of study;
- can tolerate enteral feeding including trophic feeds;
- not on steroids at randomization and no intent to initiate steroids while in trial;
- ethical approval by a human research ethics board;
- signed consent form obtained from parent/legal guardian;
- started enteral feeding within the first 7-14 days of life.
Exclusion criteria
- history of underlying disease, or condition and congenital malformation, which, in the opinion of the investigator, is likely to interfere with the normal growth and development or the evaluation of the participant;
- had undergone major surgery that required general anesthesia before randomization;
- small for gestational age, as defined as <10th percentile on a sex-specific Fenton Preterm Growth Charts;
- on fluid restriction <120 ml/kg/day;
- have periventricular/intraventricular hemorrhage;
- with greater than Grade II NEC, confirmed sepsis, renal failure/insufficiency, maternal incapacity (including maternal cocaine or alcohol abuse during pregnancy or at time of enrollment), maternal or infant HIV positivity, asphyxia resulting in severe and permanent neurologic damage, seizures, or uncontrolled systemic infection at the time of enrollment;
- participation in another clinical study that has not been approved by the sponsor;
- have a maternal history with known adverse effects on the fetus and/or the newborn infant, such as diabetes, active tuberculosis, perinatal infection, or substance abuse;
- 5-minute Apgar score <4 [appearance, pulse, grimace (distorted expression), activity and respiration]
- reasonable likelihood of transfer to another NICU where human research ethics approval had not been secured.
3.5.5 Sample size
The sample size calculation should be based on the primary outcome, with methodologies/criteria specially designed for a non-inferiority or superiority trial. Determination of the appropriate sample size depends on various factors including the choice of non-inferiority or superiority margins and statistical power.
The choice of MNI, which is based on clinically important difference in weight gain rate over the study duration, should be determined based on statistical evidences (e.g., findings of systematic review and meta-analysis of recent growth and tolerance studies in preterm infants) and clinical judgment (26). The standard deviation of weight gain rate taking into account variability in sex, birth weight, gestational age over the study duration should be determined based on historical data (e.g., data regarding weight gain rate in preterm infants derived from several clinical trial of growth in preterm infants).
The sample should be sufficiently large to have a high probability (power) of detecting a statistically significant difference of at least the size of the margin, if such a difference exists across groups, using the planned analysis.
As discussed above, the MNI of -1.1 g/kg/day and minimal clinically important difference of 1.1 g/kg/day of weight gain rate for preterm infants, over a 28- to 31-days study interval, beginning between 7 and 21 days of life, are considered the minimal clinically acceptable differences of weight gain rate between the test and control groups. Recent studies also suggest that the standard deviation of weight gain rate for a 4-weeks study period, starting within 21 days of life, is about 4.2 g/kg/day (12,28,33).
Therefore, it is recommended that for a non-inferiority trial, sample size should be calculated to test if the test preterm infant formula or fortified human milk is inferior to the control, with the non-inferiority margin of -1.1 g/day and the common standard deviation of 4.2 g/kg/day. Most often, α would be specified between 0.025 and 0.05, and β between10% and 20% and the anticipated dropout rate between 30% and 35%, respectively.
For a superiority trial of growth and tolerance in preterm infants, sample size should be calculated to test whether or not the experimental preterm infant formula or fortified human milk is superior to the concurrent approved control with sufficient power to detect the minimum clinically important difference of 1.1 g/kg/day, assuming the common standard deviation is 4.2 g/kg/day. The α, β and anticipated dropout rate would often be specified as for the non-inferiority trial.
3.5.6 Intervention
Precise details of the intervention intended for each group should be reported. It is recommended that subjects be assigned in a balanced ratio to the following groups:
- Test product
The test product should be the one proposed for the Canadian market, i.e., with the same composition, nutritional profile and manufacturing process and that is also in compliance with the nutrient requirements of the FDR (for preterm infant formula). It should also be in line with or slightly exceed the international expert nutrients recommendations for preterm infant (Appendix 3).
- Control product
A concurrent control should be included in the study. It is recommended that a Canadian commercial preterm infant formula or human milk fortifier + preterm human milk be used as a control.
The nutritional profile of the combination of the human milk fortifier + preterm human milk for each group should be included in the study report and submitted in the premarket submission. It is recommended that the nutritional profile be based on the analysis of 2-3 samples of this combination at the point of full enteral feeding of fortified human milk.
If a sufficient quantity of mother's own milk is not available, term donor milk may be used with a human milk fortifier, but mother's own preterm milk is recommended (40), please see Appendix 4 and 5 for the composition of preterm human milk and the different volume and energy used by experts in their determination of nutrient recommendations.
In the case of a preterm infant formula or a human milk fortifier with an added new ingredient, the test and control products should be identical except for the new ingredient.
- Comparison
The protocol should identify and describe any differences in composition and nutritional profiles, including the energy density, protein source of the test and control products.
3.5.7 Study duration
Subjects should exclusively (i.e. more than 80% of daily intake) receive the assigned preterm infant formula or fortified human milk for at least 4 weeks or until hospital discharge. Longer-term follow-up (i.e. up to 36 – 40 weeks corrected age) is strongly recommended.
3.5.8 Assignment
Participants' allocation to the treatment or control groups should be randomized using concealed allocation and following the CONSORT guidelines (14,19). Randomization reduces selection bias by controlling for known and unknown confounding factors in the study population, when well conducted and concealed. The randomization process includes the following aspects (14,39):
- Sequence generation
Describe the method used to generate the random allocation sequence. Adequate methods include the use of a random-number table or a computerized random number generator (19). Subjects' randomization should be stratified based on birth weight category, center (in multicenter trial) and sex since boys have a different growth rate than girls.
- Allocation concealment
The sequence of random allocation should be concealed until recruitment is complete, irrevocable, and interventions have been assigned. For example, treatment allocation could be concealed by using computerized randomization and allocating tool or enclosing assignments in sequentially numbered, opaque, sealed containers or sealed and stapled opaque envelopes (19).
Good allocation concealment mechanisms, which incorporate external involvement are recommended, for example, the use of a pharmacy or central telephone randomization system are two common techniques (19).
- Randomization implementation
The protocol should include detailed information on all personnel who are responsible for generating the random sequence, enrolling participants and allocating participants to each study arm (19). Any deviation from random allocation should be described in the study report.
3.5.9 Blinding
Study participants (parents/caregivers), investigators, outcome assessors and all other study personnel should be blinded to treatment assignment. The test and control products should be identical in appearance in case of formula, for example, packaging, texture, taste and smell (39). Detailed information should be given on how products are labelled (e.g., by individual subject codes), who has access to the product codes, whether there are any pre‐defined circumstances in which the blinding could be broken and who from the team of investigators would be un-blinded in case of such a need. In addition, blinding should be maintained until the study is completed. Outcomes assessors should be blinded at all times.
3.5.10 Data collection and outcomes
3.5.10.1 Collection schedule
Describe the data collection schedule. Subjects in the test and control groups should be enrolled and followed in parallel. The NICUs' clinical feeding standard protocols should be provided.
Standardized procedures and calibrated instruments should be used in collecting measurement data. Study personnel training, and quality assurance procedures, should be in place to ensure measurements are as precise and accurate as possible across participants, study sites, and time of data recording. In cases where incomplete data are collected, the extent and impact of the missing data should be described in the study report and the approach chosen to handle the missing data should be reported.
3.5.10.2 Collection of basic demographic dataset
All infants included in the clinical study should be well characterized, especially with regard to factors that might affect the planned outcomes. In order to allow a comprehensive scientific assessment of the study, the following information should be provided:
Infant data
- infant sex;
- method of delivery (vaginal, C-section);
- single birth, twins or triplet;
- gestational age at birth and baseline;
- birth weight per gestation (appropriate for gestational age etc.);
- anthropometry at baseline including body weight, length and head circumference in absolute values and z-scores, together with a documentation of the growth standard used to calculate the z-scores. Please refer to the following links to guide on measuring weight, length, and head circumference of infants: http://depts.washington.edu/growing/Assess/Anthro.htm;
- 5 minutes Apgar score [appearance, pulse, grimace (distorted expression), activity and respiration];
- chronological age at recruitment, randomization and baseline (that is, at the start of the intervention) and at each assessment point;
- race and/or ethnicity;
- concurrent medications use (caffeine, steroids, etc.);
- underlying clinical conditions, acuity and severity;
- antenatal steroids;
- age in days when enteral feeding started;
- age when started fortification;
- growth charts used.
Maternal data
- maternal age, education and body mass index (BMI);
- history of smoking during pregnancy or at randomization;
- maternal alcohol, drugs, or substances abuse during pregnancy and at randomization;
- mother's parity;
- maternal history with known adverse effects on the fetus and/or the newborn infant, such as diabetes, active tuberculosis or perinatal infection.
Paternal data
- paternal age and education;
- parental heights and weights, measured if possible;
- socioeconomic status.
3.5.10.3 Collection of intake data
Proper systems should be in place to assess compliance with the actual daily intakes of the assigned formula or fortified human milk during the study. This data is important for the interpretation of trial results.
3.5.10.4 Outcome measurements
The following outcome parameters must be measured at pre-determined time-points over the study duration. Any methods used to enhance the quality of measurements (e.g., multiple observations, training of assessors) should be reported.
Primary outcome
- Rate of weight gain over the course of the study expressed as g/kg/day calculated using either exponential model or 2-point model for the average weight gain calculation (9,10,11,22) between study day 0 and end of study.
Secondary outcomes
The following growth and safety outcomes should be compared between the test and control groups:
- weekly rates of length (cm/week) and head circumference gains (cm/week) between study Day 0 and the end of the study;
- attained weight, length and head circumference at baseline and each study visit for each individual and for each group stratified by sex, birth weight and/or gestational age category plotted on the Fenton Preterm Growth Charts (21);
- group calculated z-scores and changes in z-scores and their 95% confidence intervals for weight-, length-, and head circumference-for-age and weight-for length, at birth, baseline, each study visit and at discharge using Fenton Preterm Growth Charts (21) by sex, birth weight and/or gestational age category;
- time to regain birth weight;
- time to reach 1800 -2000 g body weight which is considered an important milestone in this area of research (potential time for hospital discharge based on a case-by case);
- time to start fortification;
- time to reach full enteral feeding, 150 ml/kg/day;
- incidence of weight loss of more than 2 z-scores relative to the Fenton Preterm Growth Charts (21) at 36 weeks or at discharge (5);
- number of infants discharged as small for gestational age defined as body weight <10th percentile of a given gestational age relative to the Fenton Preterm Growth Charts (21);
- duration of total parenteral nutrition (TPN) (days);
- duration of enteral nutrition;
- duration on the study products;
- duration of hospital stay (days);
- proportion of infants with ≥ 1 episode of feed interruption lasting ≥ 12 hours;
- incidence and daily records of adverse events and serious adverse events, e.g., mortality, renal failure/insufficiency, sepsis, NEC, etc. reported, verified and assessed by blinded health care professionals;
- average daily intake volume of preterm infant formula or fortified human milk (mother's own milk or donor milk), calories, protein, modular supplements and vitamins and minerals supplementation;
- daily records of tolerance data and its physicians' blinded appraisal (withhold of enteral feeding, gastric residuals, abdominal distention, vomiting, colic, fussiness, cramps, sleeping patterns, regurgitation and stool characteristics, hospital stay, etc.);
- frequencies and severity of adverse events and serious adverse events and total number of participants with these events;
- any planned biochemical tests at baseline, mid point and end of the study, e.g., creatinine, prealbumin, BUN, serum alkaline phosphatase, serum electrolytes, calcium, phosphorus, magnesium, haemoglobin, hematocrit, serum iron, total iron binding capacity, blood gases, blood pH, urinary pH, etc. The need for such tests depends on the nature of the formulation and would be determined on a case-by-case basis;
- days of intermittent positive pressure ventilation (IPPV);
- days of continuous positive airway pressure (CPAP);
- days of mechanical ventilation;
- days on supplementary oxygen, probiotics, which probiotic provided and when
- proportion of intake from own mother or donor milk;
- number of infants with NEC, using Bell's criteria > stage II;
- number of suspected sepsis treated with antibiotics;
- number of proven sepsis treated with antibiotics;
- number of infants being discharged home or transferred and age if yes
- number of non-compliance/withdrawn.
Note: for longer term follow-up studies The World Health Organization Child Growth Standards (2006) (2) should be used to assess growth over a study duration longer than 50 weeks postmenstrual age.
Post-marketing surveillance
A post-marketing (also known as in-marketing) surveillance plan may be considered to support long-term safety (41). Post-marketing surveillance consists of:
- a monitoring component which includes procedures to monitor for adverse effects once the product has been put on the market;
- a follow-up component which focuses on potential long-term or delayed effects following the period of maximum usage of the product. The length of the follow-up should be determined on a case-by-case basis;
- a biannual or annual written report from the petitioner's medical and scientific committee summarizing the results of its literature review, particularly, those pertaining to the safety and long-term follow-up of any added new ingredient (e.g., lutein);
- growth and developmental outcomes assessment (determined on case-by case basis).
3.5.11 Adverse events
Accurate and clear information on adverse events and side effects (unexpected) occurring during the study or during a specified follow-up period must be carefully described and reported whether or not they are immediately associated with the study treatment (42).
The protocol to ascertain possible adverse events and side effects should be defined a priori and reported. This should include the duration and frequency of monitoring, a valid and recognized case definition for common or anticipated adverse events with standardized criteria, checklists or validated scales/scores for diagnosis, where appropriate. The person(s) who detected or confirmed the adverse event must also be documented (investigator, medical records, medical professional, clinical personnel, sponsor). When pertinent, the method for dealing with and counting recurrent events should be documented. Any trial stoppage guidelines related to adverse events should be defined and described.
In case of adverse events, the following information should be documented in the medical records and study report and confirmed by medical professionals for each case, as appropriate:
- subject number, date and duration of event;
- description of the event;
- severity (mild, moderate, severe);
- seriousness and frequency;
- potential attribution to the study feeding;
- action taken and details of any medications used;
- participant withdrawal following adverse event.
3.5.12 Stopping rules, discontinuation, withdrawals and dropouts
The protocol should provide details of the procedure to monitor serious adverse events and should indicate the stopping rules, discontinuation criteria or withdrawal criteria for individual subjects, parts of trial and entire trial, and the plan to follow-up withdrawals and dropout (42).
Mechanisms should be in place to reduce withdrawals from the study as much as possible for reasons other than adverse events. For all participants who withdraw from the study prior to the study completion, the principal investigator should make reasonable attempts to follow up to determine the reasons for dropping-out. The following details of subjects who fail to comply, or who withdraw or dropout should be carefully captured and reported:
- detailed description of participants who have failed to comply, or who withdrew or dropped-out;
- description of reason(s) for withdrawal or dropping-out from study;
- date and age when stopped participating as per protocol;
- details of intakes (parenteral, preterm infant formula, human milk, fortified human milk and modular supplements) before dropping out or withdrawal;
- length of time subject was in the study before dropping out;
- reason(s) for non-compliance;
- postnatal age at withdrawal or dropping-out from the study.
3.5.13 Quality control procedures
The protocol should provide a description of the quality control procedures with written standard operating procedures (SOP) to ensure that the trial is conducted and data is generated, documented (recorded), and reported in compliance with the protocol, good clinical practice (GCP), and the applicable regulatory requirement(s) (43). The SOP also ensures that the recruitment into the trial is conducted properly to minimize potential sources of "selection bias" and all outcome measurements collected during the study are accurate, reproducible, valid and reliable.
3.5.14 Independent Data Monitoring Committee (IDMC)
The sponsor should establish an IDMC to periodically assess the progress of the clinical trial, safety data and efficacy parameters, and to recommend to the sponsor whether to continue the trial, modify or terminate the trial. The IDMC should have written SOPs and maintain records of all its meetings, including interim results. The committee should include a biostatistician with knowledge of clinical trial design and analysis; clinical trial scientists knowledgeable in the field; and a neonatologist (18,43).
3.5.15 Protocol violation
Protocol violations should be reported in the study report and the registry should be updated. The study report should document the assessment of the magnitude of these violations and how much influence they may have on the interpretation of the results (18,44).
3.5.16 Protocol amendment
Protocol deviations or any significant amendments required during the study should be reported in the study report and study protocol and the registry should be updated, specifically noting that a change was made. The rationale for any significant amendment and its influence on the interpretation of the results should be assessed, reported in the study report and protocol, and the study registration updated (18).
3.5.17 Pre-planned statistical analyses
Overall, the statistical analysis and methods used must be in line with generally accepted scientific principles (45).
The protocol must clearly state the test hypotheses (e.g., non-inferiority or superiority test hypothesis) and/or the treatment effects, which are to be estimated in order to satisfy the primary and secondary objectives of the trial. The statistical methods to be used to accomplish these tasks must be clearly described for the primary as well as the secondary analyses and the underlying statistical model must be made clear a priori. A description must be given of any intentions to use baseline data to improve precision or to adjust estimates for potential baseline differences, for example by means of analysis of covariance.
Sample size must be based on the detection of an effect size for the primary outcome, which is both statistically significant and clinically meaningful. The statistical method chosen must be concordant with the study's design and sample size calculation conducted to ensure the planned analyses will be sufficiently powered to detect a significant difference between study groups to provide a reliable answer to the questions addressed (e.g., new test product is not inferior to the control product, or new test product is superior to the control product). The study power and the statistical significance level must be reported with the expected attrition rate, which must be accounted for in sample size calculation. Calculation of the statistical power at the end of the study is also required (23). Timing of any planned interim analyses and criteria for the termination of the trial must be indicated.
The criteria for inclusion/exclusion of subjects in the per-protocol (PP) analysis must be planned a priori in the protocol. Detailed description of reasons, method used and rationale for any potential data exclusion and/or adjustment (e.g., deleting a partial data, replacing missing values, adjusting invalid values, etc.) from the analysis must be clearly discussed and described in the study report.
Results must be provided for comparisons between the test and control groups for all outcome variables assessed. Growth patterns of the study groups must also be compared with acceptable growth references for preterm infants (21). In particular, the following information must be provided (20):
- For a non-inferiority trial, descriptive and inferential statistics (such as means, standard deviations, 95% confidence intervals and measures of statistical significance) for each measured outcome for each group at the beginning of the study and at every point of assessment throughout the study must be reported and analyzed; both per-protocol and ITT analyses must be performed (20). Non-inferiority and the robustness of the results are clearly established when both PP and ITT analyses demonstrate lack of statistically significant difference between test and control preterm infant formula or fortified human milk groups. For a superiority trial, the above statistics for the primary and secondary outcomes must be based on the ITT analysis. The PP analysis must be also performed to assess the consistency of results derived from these analyses;
- Estimates of effect size (e.g., difference in weight gain between the test and control groups) must be accompanied with its inferences (precision);
- The number of infants analysed at each time point for each analysis;
- Statistical models and covariates used in the analysis, with appropriate justification for their use;
- The presentation and interpretation of the results using confidence interval in relation to the MNI or superiority margin;
- The results of both the adjusted and unadjusted analysis;
- For each sex, birth weight and/or gestational age category, plotted mean achieved body weight, length, and head circumference and their 95% confidence intervals against postmenstrual age by study group;
- For each sex, birth weight and/or gestational age category, plotted weight, length, and head circumference-for-age, weight-for length z-scores and changes in z-scores, based on Fenton Preterm Growth Charts (21), for each study group over the study period;
- Total number and reasons for dropouts or withdrawals of infants from the study by investigators for each study group, together with an assessment/discussion of the impact of dropouts/withdrawals on the study results;
- If there is a plan to conduct a subgroup analysis or to adjust for any confounders at the analysis stage, details must be specified a priori in the protocol.
Handling of missing data
If imputation (replacing the missing values with estimates) of missing data is foreseen, information on the robustness of the assumptions made should be reported. Detailed explanations should be provided in the study report as to how such estimations or derivations were done; number and reasons of missing values in each group; what underlying assumptions were made, and the impacts of imputations on the study conclusions.
Multiple comparisons
For studies for which an adjustment for multiple comparisons is needed, the pre-planned approach towards adjusting for multiplicity should be specified.
3.6 Study results
The study results must be reported in a transparent and reproducible manner in line with the CONSORT Statement (14).
3.6.1 Participants flow chart and analysis of withdrawals and dropouts
A flow chart is strongly recommended, as it is useful in illustrating participants flow throughout the study, from recruitment to study completion. Any deviation from planned protocol should be described together with reasons. For each study arm, the following detailed information should be indicated in the flow chart:
- total number of subjects randomized, receiving intended intervention, completing the study protocol, and being analysed for the primary and secondary outcomes;
- total number of subjects withdrawing or dropping-out from the study and being excluded from the primary and secondary analyses of outcomes.
A table showing baseline demographic and clinical characteristics for each group should be included.
3.6.2 Baseline analyses
Include results of analyses comparing the socio-demographic characteristics and anthropometric measurements at birth and baseline between the test and control groups in the report.
3.6.3 Analyses of primary and secondary outcomes
For Non-inferiority Trial: include results derived from the PP and ITT analyses of the primary and secondary outcome parameters (listed under "Outcomes measurements" Section 3.5.10.4) between the test group vs. control group in the final report.
For Superiority Trial: include results derived from the ITT analysis of the primary and secondary outcome parameters between the test group vs. control group in the final report.
In particular, clearly report the following findings:
- number of participants analysed at each predefined time point in each of the PP and ITT analyses;
- descriptive statistics including mean, median, and its inferences for each outcome variable by sex, birth weight and/or gestational age category and study arm;
- mean z-scores and changes in z-scores for weight, length, head circumference-for- age, weight-for-length and its inferences by sex, birth weight, gestational age category and study group using Fenton Preterm Growth Charts (21);
- achieved weight, length and head circumference at each study visit and over the study duration by sex, birth weight and gestational age category and study arm;
- estimates of mean differences, their inferences and p-values in each assessed outcome across the above groups, as well as detailed discussions regarding significant differences.
Fenton Preterm Growth Charts (21) should be used to compare the study group's mean attained weight, length, and head circumference and calculated z-scores at each study visit and over the duration of the study.
- Individual growth charts
The individual anthropometric data should be plotted on the Fenton Preterm Growth Charts (21) in order to trace the growth performance of each individual. Individual growth charts must be provided in the preterm infant formula or human milk fortifier premarket submission.
- Z-scores
The individual anthropometric measurements should be transformed to z–scores based on the Fenton Preterm Growth Charts (21). These z-scores indicate the distance and direction of an observation away from the population mean (standard deviations above and below the mean).
Group z-scores: z-scores for weight, length, and head circumference-for-age and weigh-for-length for each group categorized by sex, birth weight and gestational age category should be compared and submitted in PMNs.
Fenton z-score calculators can be downloaded from www.ucalgarry.ca/fenton.
As mentioned above, in case of long-term follow-up study, the World Health Organization Child Growth Standards (2) should be used to assess growth in a study duration longer than 50 weeks postmenstrual age.
3.6.4 Analyses of adverse events
Detailed information, listed under adverse events Section 3.5.11, for subjects with incidents of adverse events should be interpreted by comparing the occurrence of adverse events in each study arm, withdrawals resulting from these events, and also by assessing the severity against the number of events. It is strongly recommended that the clinical assessment of adverse events and its attribution to the assigned treatment be conducted in a blinded manner if possible by independent medical professionals. These data should inform a balanced assessment of harms and benefits.
If further examination is required to assess the relationship between an occurrence of a serious adverse event and the intervention, all pertinent examinations or laboratory findings must be noted with their results in the CRF or attached to a follow-up file.
3.7 Discussion and conclusions
Discussion and interpretation of the study results should clearly distinguish clinical from statistical significance. The authors' conclusions should be in line with the reported study results taking the non-inferiority or superiority hypotheses into account, with the primary outcome clearly identified and prioritized. It should be indicated whether the conclusions relating to non-inferiority or superiority are based on PP or ITT analysis or both, and whether the conclusions are consistent with both analyses for non-inferiority trials. They should include a balanced assessment of the benefits and harms with emphasis on study limitations, and consider any other relevant evidence including consistency or inconsistency with other trials, sources of potential bias or imprecision, different standards of care and standard clinical protocols in NICUs, lack of or partial blinding, dangers associated with multiplicity of analyses and outcomes and generalizability (external validity, applicability of the study findings).
4.0 Ethical standard for investigators
4.1 Informed consent (International Conference on Harmonisation (ICH) E 6 (R1))
Each potential subject (parents or caregivers) must be adequately informed of the aims, methods, sources of funding, any possible conflicts of interest, institutional affiliations of the researchers, the anticipated benefits and potential risks of the study and the discomfort it may entail, post-study provisions and any other relevant aspects of the study. The potential subject must be informed of the right to refuse to participate in the study or to withdraw consent to participate at any time without reprisal (43).
The study is to be conducted in accordance with the ethical principles and rules that have their origin in the declaration of Helsinki and its subsequent amendments and should be consistent with GCP and by any applicable regulatory requirements.
The protocol, any amendments and the informed consent process should receive Institutional Review Board (IRB) approval/favourable opinion prior to initiation. The institution granting the approval should be reported. The name of any contract research organisation that has been tasked to carry out the work should be provided.
Investigational products should be manufactured, handled and stored in accordance with applicable good manufacturing practices. They should be used in accordance with the approved protocol.
4.2 Funding, sponsorship and conflict of interest
The protocol should include information regarding funding, sponsors, institutional affiliations, potential conflicts of interest, incentives for subjects and information regarding provisions for treating and/or compensating subjects who are harmed as a consequence of participation in the research study. The exact role and contribution of the funder and sponsor to the study should be disclosed (e.g., in the design, conduct, analysis and/or reporting) (18).
An investigator who is ideally independent of the infant formula industry should take overall responsibility for the conduct of the trial, planning and conduct of statistical analyses, decision to publish, reporting and interpretation of the trial findings (18).
4.3 Investigators credentials
The protocol should describe the necessary qualifications and experience of the investigators.
4.4 Long-term follow-up studies
Collecting clinical data via long-term follow-up studies (i.e. up to 20 months of corrected age) in premature infants is encouraged: it will provide more insights on growth quality and address how to balance weight gain to optimize neurodevelopmental outcomes against the risk of early-onset cardiovascular and metabolic diseases (36, 46).
The following long-term parameters could be assessed:
- developmental outcomes, i.e. Bayley scores or intelligence quotient tests, assessed usually after 20 months of corrected age;
- assessment of body composition, i.e. changes in lean mass and fat mass.
Note, in view of the advancing and evolving field of the nutritional management of preterm and low birth weight infants, this protocol will be updated in the future based on new research data.
5.0 Glossary and references
5.1 Glossary
Adjusted analysis
Usually refers to attempts to control for baseline imbalances between groups in important patient characteristic. Sometimes used to refer to adjustment of p-value to take account of multiple testing (42).
Adverse event
An adverse outcome that occurs during or after the use of a drug or other intervention but is not necessarily caused by it (42).
Adverse event grading of mild, moderate, and severe
This is commonly used in non-oncology studies. The definition of the mild, moderate, and severe may be different from one study protocol to another. The severity (intensity) of each adverse event including serious adverse events are assigned to one of the following categories:
- mild: an event that is easily tolerated by the subject, causing minimal discomfort and not interfering with everyday activities
- moderate: an event that is sufficiently discomforting to interfere with normal everyday activities
- severe: an event that prevents normal everyday activities
OR
- mild: awareness of sign or symptom, but easily tolerated
- moderate: discomfort sufficient to cause interference with normal activities
- severe: incapacitating, with inability to perform normal activities (42)
Adverse effect
Is an adverse event for which the causal relation between the intervention and the event is at least a reasonable possibility by it (42).
Adverse reaction
Events for which a causality link to the intervention is well established and strong enough (sensitive and specific) to warrant attribution of the event to the intervention (42).
Allocation concealment
Is a technique used to ensure an unpredictable sequence of assignments. It is a critical process that prevents foreknowledge of treatment assignment and thus shields those who enroll participants from being influenced by this knowledge. The decision to accept or reject a participant should be made, and informed consent should be obtained from the participant, in ignorance of the next assignment in the sequence (47).
Allocation sequence
A list of interventions, randomly ordered, used to assign sequentially enrolled participants to intervention groups (42).
Apgar score
An objective score of the condition of a baby after birth. This score is determined by scoring the heart rate, respiratory effort, muscle tone, skin color, and response to a catheter in the nostril. Each of these objective signs receives 0, 1, or 2 points. An Apgar score of 10 means an infant is in the best possible condition. The Apgar score is done routinely 60 seconds after the birth of the infant. A child with a score of 0 to 3 needs immediate resuscitation. The Apgar score is often repeated 5 minutes after birth, and in the event of a difficult resuscitation, the Apgar score may be done again at 10, 15, and 20 minutes.
Attrition
The loss of participants during the course of a study (also called loss to follow up). Participants that are lost during the study are often call dropouts (48).
Bias
Systematic distortion of the estimated intervention effect away from the "truth" caused by inadequacies in the design, conduct, or analysis of a trial (42).
Blinding
The process of preventing those involved in a trial from knowing to which comparison group a particular participant belongs. It is the practice of keeping the trial participants, care providers, data collectors, and sometimes those analyzing data unaware of which intervention is being administered to which participant (42).
Clinical trial
An experiment to compare the effects of two or more healthcare interventions. Clinical trial is an umbrella term for a variety of designs of healthcare trials, including uncontrolled trials, controlled trials, and randomized controlled trials (48).
Confidence interval
A measure of the precision of an estimated value. The interval represents the range of values, consistent with the data that is believed to encompass the "true" value with high probability (usually 95%). The confidence interval is expressed in the same units as the estimate. Wider intervals indicate lower precision; narrow intervals indicate greater precision (49).
Confounder
A factor that is associated with both an intervention (or exposure) and the outcome of interest (48).
Control
In a controlled trial: A participant in the arm that acts as a comparator for one or more experimental interventions. Controls may receive placebo, no treatment, standard treatment, or an active intervention, such as a standard drug;
In statistics: To adjust for, or take into account, extraneous influences or observations (48).
Effect size
A generic term for the estimate of effect of treatment for a study;
A dimensionless measure of effect that is typically used for continuous data when different scales (e.g., for measuring pain) are used to measure an outcome and is usually defined as the difference in means between the intervention and control groups divided by the standard deviation of the control or both groups (48).
Efficacy
The extent to which an intervention produces a beneficial result under ideal conditions. Clinical trials that assess efficacy are sometimes called explanatory trials and are restricted to participants who fully co-operate (48).
Efficacy vs. effectiveness
Intervention studies can be placed on a continuum, with a progression from efficacy trials to effectiveness trials. Efficacy can be defined as the performance of an intervention under ideal and controlled circumstances, whereas effectiveness refers to its performance under 'real-world' conditions (50).
External validity
The extent to which the results of a trial provide a correct basis for generalization to other circumstances (42).
Generalisability, generalisation
The extent to which the findings of a clinical trial can be reliably extrapolated from the subjects who participated in the trial to a broader patient population and a broader range of clinical settings (45).
Hypothesis test
A statistical procedure to determine whether to reject a null hypothesis on the basis of the observed data (48).
Imputation
A procedure for entering a value for a specific data item where the response is missing or unusable. This is done by changing some of the responses or assigning values when they are missing on the record being edited to ensure that estimates are of high quality and that a plausible, internally consistent record is created (51).
Intention-to-treat analysis
A strategy for analysing data from a randomised controlled trial. All participants are included in the arm to which they were allocated, whether or not they received (or completed) the intervention given to that arm. ITT analysis prevents bias caused by the loss of participants, which may disrupt the baseline equivalence established by randomisation and which may reflect non-adherence to the protocol. The term is often misused in trial publications when some participants were excluded (48).
Mean
An average value, calculated by adding all the observations and dividing by the number of observations (48).
Multicenter trial
A clinical trial conducted according to a single protocol but at more than one site, and therefore, carried out by more than one investigator (45).
Multiplicity
The proliferation of possible comparisons in a trial. Common sources of multiplicity are multiple outcomes, outcomes assessed at several time points after the intervention, subgroup analyses, or multiple intervention groups (49).
Non-inferiority margin (MNI)
The margin is the largest difference that can be judged as being clinically acceptable and should be smaller than differences observed in superiority trials of the active comparator (45).
Non-inferiority trial
A trial with the primary objective of showing that the response to the investigational product is not clinically inferior to a comparative agent (active or a placebo control) [26], a one-sided version of an equivalence trial (48).
Null hypothesis
In simplest terms, the null hypothesis states that the factor of interest (e.g. treatment) has no impact on outcome (e.g., risk of death) (48).
P-value
The probability (ranging from almost zero to one) that the results observed in a study (or results more extreme) could have occurred by chance if in reality the null hypothesis was true.
Per-protocol
An analysis of the subset of participants from a randomized controlled trial who complied with the protocol sufficiently to ensure that their data would be likely to exhibit the effect of treatment. This subset may be defined after considering exposure to treatment, availability of measurements and absence of major protocol violations. The per protocol analysis strategy may be subject to bias as the reasons for non-compliance may be related to treatment (48).
Power
The probability that a trial will detect as statistically significant an intervention effect of a specified size. The pretrial size is often chosen to give the trial the desired power (45).
Precision [In statistics]
A measure of the likelihood of random errors in the results of a study, meta-analysis or measurement. The greater the precision, the less random error. Confidence intervals around the estimate of effect from each study are one way of expressing precision, with a narrower confidence interval meaning more precision (48).
Primary outcome
The outcome deemed of greatest importance (48).
Protocol deviation
A protocol deviation occurs when, without significant consequences, the activities on a study diverge from the Institutional Review Board-approved protocol, for example, missing a visit window because the subject is traveling. Not as serious as a protocol violation (44).
Protocol violation
A divergence from the protocol that materially: reduces the quality or completeness of the data,
makes the Informed Consent Form inaccurate, or impacts a subject's safety, rights, or welfare (44).
Safety
Substantive evidence of an absence of harm. The term is often misused when there is simply absence of evidence of harm (42).
Selection bias
Systematic error in creating intervention groups, causing them to differ with respect to prognosis. That is, the groups differ in measured or unmeasured baseline characteristics because of the way in which participants were selected for the study or assigned to their study groups. The term is also used to mean that the participants are not representative of the population of all possible participants (45).
Serious adverse event (in the context of a clinical trial)
When in the view of either the investigator or sponsor it results in any of the following outcomes: Death, a life threatening adverse event, inpatient hospitalization or prolongation of existing hospitalization, a persistence or significant incapacity or substantial disruption of the ability to conduct normal life functions (43).
Side effect
Is an adverse event for which the causal relation between the intervention and the event is at least a reasonable possibility by it (42).
Standard deviation
A measure of the spread or dispersion of a set of observations, calculated as the average difference from the mean value in the sample (48).
Standard (Std) fortification
This is the most widely used fortification method. The standard practice is to add a fixed amount of multi-nutrient fortifier per 100 mL of HM to achieve the recommended nutrient intakes. This fixed amount has been calculated and determined by the manufacturer assuming a fixed protein content for all milk samples without considering intra-, inter-individual and temporal variations. Standard fortification is initiated usually when the fed milk volume is 50–100 mL/kg. Milan EMBA/ESPGHAN/AAP Joint Meeting Consensus recommends fortifying HM for preterm infants with a birthweight <1,800 g (53).
Superiority trial
A trial with the primary objective of showing that the response to the investigational product is superior to a comparative agent (active or placebo control) (45).
Trophic feeding or minimal enteral nutrition
Small volume enteral milk feed in the first 24 hours.
Type I error
A conclusion that a treatment works, when it actually does not work. The risk of a Type I error is often called alpha. In a statistical test, it describes the chance of rejecting the null hypothesis when it is in fact true (also called false positive) (48).
Type II error
A conclusion that there is no evidence that a treatment works, when it actually does work. The risk of a Type II error is often called beta. In a statistical test, it describes the chance of not rejecting the null hypothesis when it is in fact false. The risk of a Type II error decreases as the number of participants in a study increases (also called false negative) (48).
Z-score
Is the deviation of the value for an individual from the mean value of the reference population divided by the standard deviation for the reference population (52).
5.2 References
- American Academy of Pediatrics, Committee on Fetus and Newborn. (2004). Age terminology during the perinatal period. Pediatrics, 114 (5), 1362-1364.
- World Health Organization (WHO). (2006). Optimal feeding of low-birth-weight infants. [Online]. Available: https://apps.who.int/iris/bitstream/handle/10665/43602/9789241595094_eng.pdf;jsessionid=80736F92F581729831081B1CA71D704B?sequence=1
- World Health Organization (WHO). (2018). Preterm Birth: Fact sheet. [Online]. Available: https://www.who.int/news-room/fact-sheets/detail/preterm-birth.
- Sharma D, Shastri S, Sharma P. (2016). Intrauterine Growth Restriction: Antenatal and Postnatal Aspects. Clinical Medicine Insights: Pediatrics. 10, 67-83.
- Goldberg D, Becker PJ, Brigham K, Carlson S, Fleck L, Gollins L, Sandrock M, Fullmer M, Van Poots HA. (2018). Identifying Malnutrition in Preterm and Neonatal Populations: Recommended Indicators. J Acad Nutr Diet. 118(9),1574-1582.
- Goldenberg R, Culhane JF, Iams JD, Romero R. (2008). Epidemiology and causes of preterm birth. The Lancet, 371(9606), 75-84.
- Malhotra N, Puri R, Malhotra J, Chervenak FA, Kurjak A (eds). (2012). Donald School Manual of Practical Problems in Obstetrics, Jaypee Brothers Medical Publishers.
- Groh-Wargo S, Thompson M, Cox H (eds). (2000). Nutritional Care for High-Risk Newborns. Precept Press, Inc., Chicago, IL.
- Cormack BE, Embleton ND, van Goudoever JB, Hay WW Jr, Bloomfield FH. (2016). Comparing apples with apples: it is time for standardized reporting of neonatal nutrition and growth studies. Pediatr Res. 79(6):810-20.
- Patel AL, Engstrom JL, Meier PP, Kimura RE. (2005). Accuracy of methods for calculating postnatal growth velocity for extremely low birth weight infants. Pediatrics. 116(6):1466-73.
- Patel AL, Engstrom JL, Meier PP, Jegier BJ, Kimura RE. (2009). Calculating postnatal growth velocity in very low birth weight (VLBW) premature infants. J Perinatol. 29(9):618-22.
- Fenton TR, Senterre T, Griffin IJ. (2019). Time interval for preterm infant weight gain velocity calculation precision. Arch Dis Child Fetal Neonatal Ed. 104(2):F218-F219.
- Fenton TR, Griffin IJ, Hoyos A, Groh-Wargo S, Anderson D, Ehrenkranz A, Senterre T. (2019). Accuracy of preterm infant weight gain velocity calculations vary depending on method used and infant age at time of measurement. Pediatr Res., 85, 650-654.
- Schulz KF, Altman DG, Moher D; CONSORT Group. (2010). CONSORT 2010 Statement: Updated guidelines for reporting parallel group randomised trials. J Clin Epidemiol. 63(8), 834-40.
- World Health Organization (WHO). (2021). WHO primary registries. [Online]. Available: https://www.who.int/clinical-trials-registry-platform/network/primary-registries.
- World Health Organization. (2009). WHO Registry Criteria (Version 2.1, April 2009). [Online]. Available: https://www.who.int/clinical-trials-registry-platform/network/registry-criteria.
- Boutron I, Altman DG, Moher D, Schulz KF, Ravaud P; CONSORT NPT Group. (2017). CONSORT Statement for Randomized Trials of Nonpharmacologic Treatments: A 2017 Update and a CONSORT Extension for Nonpharmacologic Trial Abstracts. Ann Intern Med. 67(1):40-47.
- Jarrold K, Helfer B, Eskander M, Crawley H, Trabulsi J, Caulfield LE, Duffy G, Garcia-Larsen V, Hayward D, Hyde M, Jeffries S, Knip M, Leonardi-Bee J, Loder E, Lodge CJ, Lowe AJ, McGuire W, Osborn D and Przyrembel H. (2020). Guidance for the conduct and reporting of clinical trials of breast milk substitutes. Jama, 174(9):874-881.
- Piaggio G, Elbourne DR, Pocock SJ, Evans SJ, Altman DG; CONSORT Group. (2012). Reporting of noninferiority and equivalence randomized trials: extension of the CONSORT 2010 statement. JAMA. 308(24), 2594-604.
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies). (2017). Scientific and technical guidance for the preperation and presentation of an application for authorisation of an infant and/or follow-on formula manufacturer from protein hydrolysates. EFSA Journal, 15(5), 4779.
- Fenton TR and Kim J. (2013). A systematic review and meta-analysis to revise the Fenton growth chart for preterm infants. BMC Pediatrics, 13(59).
- Fenton TR, Anderson D, Groh-Wargo S, Hoyos A, Ehrenkranz RA, Senterre T. (2018). An attempt to standardize the calculation of growth velocity of preterm infants—evaluation of practical bedside methods. The Journal of Pediatrics, 196,77-83.
- Food and Drug Adminstration. (2014). Code of Federal Regulations Title 21, Chapter I, Subchapter B, Part 106, 106.96 and 106.121. [Online]. Available: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=106
- Kleinman R. (2019). Pediatric Nutrition, 8th Edition, American Academy of Pediatrics.
- Viswanathan S, McNelis K, Super D, Einstadter D, Groh-Wargo S, Collin M. (2015). Standardized Slow Enteral Feeding Protocol and the Incidence of Necrotizing Enterocolitis in Extremely Low Birth Weight Infants. JPEN J Parenter Enteral Nutr, 39(6):644-54.
- U.S. Food and Drug Administration. (2016). Guidance Document: Non-Inferiority Clinical Trials," 2016. [Online]. Available: https://www.fda.gov/media/78504/download.
- Rigo J, Hascoët JM, Billeaud C, Picaud JC, Mosca F, Rubio A, Saliba E, Radkë M, Simeoni U, Guillois B, de Halleux V, Jaeger J, Ameye L, Hays NP, Spalinger J. (2017). Growth and Nutritional Biomarkers of Preterm Infants Fed a New Powdered Human Milk Fortifier: A Randomized Trial. J Pediatr Gastroenterol Nutr. 65(4):e83-e93.
- Hair A, Blanco CL, Moreira AG, Hawthorne KM, Lee M, Rechtman DJ, Abrams S. (2014). Randomized trial of human milk cream as a supplement to standard fortification of an exclusive human milk-based diet in infants 750-1250 g birth weight. J Pediatr., 1655(5), 915-20.
- Kim JH, Chan G, Schanler R, Groh-Wargo S, Bloom B, Dimmit R, Williams L, Baggs G, Barrett-Reis B. (2015). Growth and Tolerance of Preterm Infants Fed a New Extensively Hydrolyzed Liquid Human Milk Fortifier. J Pediatr Gastroenterol Nutr., 61(6), 65-71.
- Lucas A, Fewtrell MS, Morley R, Lucas PJ, Baker BA, Lister G, Bishop NJ. (1996). Randomized outcome trial of human milk fortification and developmental outcome in preterm infants. Am J Clin Nutr., 64(2), 142-51.
- Moya F, Sisk P, Walsh KR, Berseth CL. (2012). A new liquid human milk fortifier and linear growth in preterm infants. Pediatrics, 130(4), e928-35.
- Schanler R, Groh-Wargo SL, Barrett-Reis B, White RD, Ahmad K, Oliver J, Baggs G, Williams L, Adamkin D. (2018). Improved Outcomes in Preterm Infants Fed a Nonacidified Liquid Human Milk Fortifier: A Prospective Randomized Clinical Trial. J Pediatr., 202, 31-37.
- Chen L and Nguyen L. Meta-Analysis for non-inferiority margins of weigh gains between formula-fed and breast-fed infant. manuscript in preparation.
- Quigley M and McGuire W. (2018). Formula versus donor breast milk for feeding preterm or low birth weight infants. Cochrane Database Syst Rev., 6(6).
- Brown J, Lin L, Embleton ND, Harding JE, McGuire W. (2020). Multi-nutrient fortification of human milk for preterm infants., Cochrane Database of Systematic Reviews, 6
- Tyson J and Ahn C. (2002). Evaluation of the growth of preterm or low birth weight infants. Food Advisory Committee on Infant Formula, Food and Drug Administration.
- Schumi J and Wittes J. (2011). Through the looking glass: understanding non-inferiority. Trials, 12(106), 1-12.
- The European Agency for the Evaluation of Medicinal Products Evaluation of Medicines for Human Use. (2000). Points to consider on switching between superiority and non-inferiority. [Online]. Available: https://www.ema.europa.eu/en/documents/scientific-guideline/points-consider-switching-between-superiority-non-inferiority_en.pdf.
- Moher D, Hopewell S, Schulz KF, Montori V, Gøtzsche PC, Devereaux PJ, Elbourne D, Egger M, Altman DG. (2010). CONSORT 2010 Explanation and Elaboration: updated guidelines for reporting parallel group randomised trials. Journal of Clinical Epidemiology, 8,63.
- Arslanoglu, S., Boquien, C. Y., King, C., Lamireau, D., Tonetto, P., Barnett, D., Bertino, E., Gaya, A., Gebauer, C., Grovslien, A., Moro, G. E., Weaver, G., Wesolowska, A. M., & Picaud, J. C. (2019). Fortification of Human Milk for Preterm Infants: Update and Recommendations of the European Milk Bank Association (EMBA) Working Group on Human Milk Fortification. Frontiers in Pediatrics, 7(76).
- Institute of Medicine (US) Committee on the Evaluation of the Addition of Ingredients New to Infant Formula. (2004). Infant formula: evaluating the safety of new ingredients. Washington (DC). National Academies Press.
- Ioannidis JPA, Evans SJW, Gøtzsche PC, O'Neill RT, Altman DG, Schulz K, Moher D for the CONSORT group. (2004). Better reporting of harms in randomized trials: an extension of the CONSORT statement. Ann Intern Med, 141(10), 781-8.
- International Conference on Harmonisation (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use. (2016). ICH harmonised tripartite guideline for good clinical practice E6 (R2). [Online]. Available: https://database.ich.org/sites/default/files/E6_R2_Addendum.pdf
- Bhatt. (2012). Protocol deviation and violation. Perspectives in Clinical Research, 3(3), 117.
- International Conference on Harmonisation (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use. (1998). ICH Harmonised Tripartite Guideline, Statistical Principles For Clincal Trials E9. [Online]. Available: https://database.ich.org/sites/default/files/E9_Guideline.pdf.
- Raiten DJ, Steiber AL, Carlson SE, Griffin I, Anderson D, Hay WW Jr, Robins S, Neu J, Georgieff MK, Groh-Wargo S, Fenton TR; Pre-B Consultative Working Groups. (2016). Working group reports: evaluation of the evidence to support practice guidelines for nutritional care of preterm infants-the Pre-B Project. Am J Clin Nutr. 103(2):648S-78S
- Deng C. (2018). Grading the severity of AEs and its impact on AE reporting. [Online]. Available: http://onbiostatistics.blogspot.com/2018/05/grading-severity-of-aes-and-its-impact.html.
- Cochrane Collaboration. (2019). Glossary of Cochrane Terms [Online]. Available: https://epoc.cochrane.org/sites/epoc.cochrane.org/files/public/uploads/SURE-Guides-v2.1/Collectedfiles/source/glossary.html.
- Altman DG, Schulz KF, Moher D, Egger M, Davidoff F, Elbourne PC, GØtzsche PC, Lang T for the CONSORT Group. (2001). The revised CONSORT statement for reporting randomized trials: explanation and elaboration. Annals of internal medicine, 134(8), 663-694.
- Singal AG, Higgins PDR, Waljee A. (2014). A Primer on Effectiveness and Efficacy Trials, Clin Transl Gastroenterol., 5(1),e45.
- Organisation for Economic Co-operation and Development. (2020). Economic Commission of Europe of the United Nations (UNECE) Glossary of terms on statistical data editing, conference of European statisticians, methodological material. [Online]. Available: https://stats.oecd.org/glossary/detail.asp?ID=3462.
- Centers for Disease Control, National Center for Health Statistics. [Online]. Available: https://www.cdc.gov/nchs/index.htm.
- Moro GE, Arslanoglu S, Bertino E, Corvaglia L, Montirosso R, Picaud JC, Polberger S, Schanler RJ, Steel C, van Goudoever J, Ziegler E. (2015). Human milk in feeding premature infants: consensus statement. Journal of Pediatric Gastroenterology and Nutrition, 61(1), S16-S19.
Appendix 1.CONSORT 2010 checklist of information to include when reporting a randomised trialFootnote 1
| Section/Topic | Item No | Checklist item |
|---|---|---|
| Title | 1a | Identification as a randomised trial in the title |
| Abstract | 1b | Structured summary of trial design, methods, results, and conclusions (for specific guidance see CONSORT for abstracts (21,31)) |
| Introduction | ||
| Background and objectives | 2a | Scientific background and explanation of rationale |
| 2b | Specific objectives or hypotheses | |
| Methods | ||
| Trial design | 3a | Description of trial design (such as parallel, factorial) including allocation ratio |
| 3b | Important changes to methods after trial commencement (such as eligibility criteria), with reasons | |
| Participants | 4a | Eligibility criteria for participants |
| 4b | Settings and locations where the data were collected | |
| Interventions | 5 | The interventions for each group with sufficient details to allow replication, including how and when they were actually administered |
| Outcomes | 6a | Completely defined pre-specified primary and secondary outcome measures, including how and when they were assessed |
| 6b | Any changes to trial outcomes after the trial commenced, with reasons | |
| Sample size | 7a | How sample size was determined |
| 7b | When applicable, explanation of any interim analyses and stopping guidelines | |
| Randomisation | ||
| Sequence generation | 8a | Method used to generate the random allocation sequence |
| 8b | Type of randomisation; details of any restriction (such as blocking and block size) | |
| Allocation concealment mechanism | 9 | Mechanism used to implement the random allocation sequence (such as sequentially numbered containers), describing any steps taken to conceal the sequence until interventions were assigned |
| Implementation | 10 | Who generated the random allocation sequence, who enrolled participants, and who assigned participants to interventions |
| Blinding | 11a | If done, who was blinded after assignment to interventions (e.g., participants, care providers, those assessing outcomes) and how |
| 11b | If relevant, description of the similarity of interventions | |
| Statistical methods | 12a | Statistical methods used to compare groups for primary and secondary outcomes |
| 12b | Methods for additional analyses, such as subgroup analyses and adjusted analyses | |
| Results | ||
| Participant flow (a diagram is strongly recommended) | 13a | For each group, the numbers of participants who were randomly assigned, received intended treatment, and were analysed for the primary outcome |
| 13b | For each group, losses and exclusions after randomisation, together with reasons | |
| Recruitment | 14a | Dates defining the periods of recruitment and follow-up |
| 14b | Why the trial ended or was stopped | |
| Baseline data | 15 | A table showing baseline demographic and clinical characteristics for each group |
| Numbers analysed | 16 | For each group, number of participants (denominator) included in each analysis and whether the analysis was by original assigned groups |
| Outcomes and estimation | 17a | For each primary and secondary outcome, results for each group, and the estimated effect size and its precision (such as 95% confidence interval) |
| 17b | For binary outcomes, presentation of both absolute and relative effect sizes is recommended | |
| Ancillary analyses | 18 | Results of any other analyses performed, including subgroup analyses and adjusted analyses, distinguishing pre-specified from exploratory |
| Harms | 19 | All important harms or unintended effects in each group (for specific guidance see CONSORT for harms [28]) |
| Discussion | ||
| Limitations | 20 | Trial limitations, addressing sources of potential bias, imprecision, and, if relevant, multiplicity of analyses |
| Generalisability | 21 | Generalisability (external validity, applicability) of the trial findings |
| Interpretation | 22 | Interpretation consistent with results, balancing benefits and harms, and considering other relevant evidence |
| Other information | ||
| Registration | 23 | Registration number and name of trial registry |
| Protocol | 24 | Where the full trial protocol can be accessed, if available |
| Funding | 25 | Sources of funding and other support (such as supply of drugs), role of funders |
Footnotes
- Footnote 1
-
CONSORT 2010 Statement: updated guidelines for reporting parallel group randomised trials (14),– recommend reading in conjunction with the CONSORT 2010 Explanation and Elaboration or important clarifications on all the items
Appendix 2. Biochemical markers of protein and bone status in preterm infantsFootnote 1
| Measurement | Advantages | Limitations |
|---|---|---|
| Protein status: | ||
| Blood urea nitrogen (BUN) | Low BUN is a good marker of low protein intake in enterally fed, clinically stable infants. | High BUN is not easy to interpret, since it may represent appropriate amino acid intake, low energy intake relative to protein intake, or amino acid intolerance. |
| Serum prealbumin | Half-life of approximately 2 days. A low level reflects current protein deficit. |
Inflammation or infection may decrease prealbumin levels. |
| Retinol-binding protein (RBP) | Half-life of approximately 12 h. A low level reflects current protein deficit. |
RBP levels may be also be affected by suboptimal iron, zinc, and vitamin A status. Measuring RBP is more expensive than prealbumin, providing equivalent information. |
| Serum transferrin | A complementary marker of protein status. | In iron deficiency, transferrin concentration increases regardless of nutritional status. It is seldom used. |
| Bone status: | ||
| Serum calcium | It is a poor marker of metabolic bone disease (MBD). | |
| Serum phosphate | High specificity and positive predictive value as a marker of MBD. | Low sensitivity and negative predictive value as a marker of MBD. Insufficient evidence as a reliable marker of MBD. |
| Serum alkaline phosphatase | Levels >900 U/L yield a specificity of 71% and a sensitivity of 88% as a marker of MBD | Insufficient evidence as a reliable marker of MBD. |
| Serum alkaline phosphatase plus serum phosphate | Alkaline phosphatase >900 U/L plus phosphate <1.8 mmol/L (5.6 mg/dL) yield a specificity of 70% and a sensitivity of 100% as a marker of MBD | Insufficient evidence as a reliable marker of MBD. |
| Urinary calcium and phosphate markers | Urinary calcium-creatinine ratio, phosphate concentration and tubular reabsorption of phosphate may be complementarily used in the diagnosis of MBD | Levels are dependent on whether infants are formula-fed or breastfed. |
Footnotes
- Footnote 1
-
Pereira-da-Silva, L., Virella, D., & Fusch, C. (2019). Nutritional assessment in preterm infants: a practical approach in the NICU. Nutrients, 11(9), 1999.
Appendix 3. Nutrient requirement template for preterm infant formula or fortifier with human milk (as fed) per 100 kcal
NOTE: Single value indicates a minimum recommendation. Value in a square bracket has been reported as the calculated value in respective citation.
- MACRONUTRIENTS
| Nutrient | FDR requirements | International expert recommendations | Nutritional profile | Specification | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Name | Unit | ESPGHAN (2010)Reference 1 | Tsang, et. al. (2005)Reference 2 >1000 g BW | LSRO (2002)Reference 3 | WHOReference 4 >1000 g BW (stabilization to term)Footnote a | Koletzko, et.al. (2014)Reference 5 | Min | Max | ||
| Protein | g | 1.8 – 4 | 3.2 – 4.1Footnote b | 2.6 – 3.8 | 2.5 – 3.6 | [2.5 – 3.0] | 3.2 – 4.1 | |||
| Fat | g | 3.3 – 6 | 4.4 – 6.0 | 4.1 – 6.5 | 4.4 – 5.7 | [3.8 – 5.7] | 4.4 – 6.0 | |||
| - LA | g | 0.5 | 0.350 – 1.400 | 0.462 – 1.309 | [0.350 – 1.425]Footnote c | NS | 0.350 – 1.400 | |||
| - ALA | g | NS | 0.050 | NS (1 – 4 E% per kg/day) |
0.077 – 0.228 (1.75 – 4% of total fatty acids) |
NS | 0.050 | |||
| - LA:ALA | - | NS | 5 – 15 | 5 – 15 | 6 – 16 | NS | NS | |||
| - ARA | g | NS | 0.016 – 0.039 | 0.022 | [0 – 0.034] Footnote d | NS | 0.032 – 0.041 | |||
| - DHA | g | NS | 0.011 – 0.027 | 0.016 | [0 – 0.020]Footnote e | NS | 0.050 – 0.055 | |||
| Carbohydrate | g | NS | 10.5 – 12 | 5.4 – 15.5 | 9.6 – 12.5 | [6.3 – 12.9] | 10.5 – 12 | |||
- VITAMINS AND MINERALS
| Nutrient | FDR requirements | International expert recommendations | Nutritional profile | Specification | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Name | Unit | ESPGHAN (2010)Reference 1 | Tsang, et. al. (2005)Reference 2 >1000 g BW | LSRO (2002)Reference 3 | WHOReference 4 >1000 g BW (stabilization to term)Footnote a | Koletzko, et.al. (2014)Reference 5 | Min | Max | ||
| Vitamin A | IU | 250 – 500 | 1200 – 2467 | 538 – 1364 | 680 – 1267 | 583 – 1250 | 1217 – 3333 | |||
| mcg RE | -- | [360 – 740] | [162 – 410] | [204 – 380] | [175 – 375] | [365 – 1000] | ||||
| Vitamin D | IU | 40 – 100 | 800 – 1000Footnote f per day | 115 – 364 | 75 – 270 | [400 – 800]Footnote f per day | 100 – 350 from milk only | |||
| Vitamin E | IU | 0.6 | [3.0 – 15] | 4.6 – 10.9 | [3.0 – 12] | [5.0 – 10.0] | [3.0 – 15] | |||
| mg α-TE | -- | 2 – 10 | [3.1 – 7.3] | 2 – 8 Footnote g | [3.4 – 6.7] | 2 – 10 | ||||
| Vitamin K | mcg | 8 | 4 – 25 | 6.2 – 9.1 | 4 – 25 | [6.7 – 8.3] | 4 – 25 | |||
| Vitamin C | mg | 8 | 10 – 42 | 13.8 – 21.8 | 8.3 – 37 | [5 – 8] | 18 – 50 | |||
| Thiamine | mcg | 40 | 125 – 275 | 138 – 218 | 30 – 250 | [33 – 42] | 127 – 273 | |||
| Riboflavin | mcg | 60 | 180 – 365 | 192 – 327 | 80 – 620 | [300 – 383] | 181 – 364 | |||
| Vitamin B6 | mcg | 35 | 41 – 273 | 115 – 191 | 30 – 250 | [15]Footnote h | 45 – 273 | |||
| Vitamin B12 | mcg | 0.15 | 0.08 – 0.7 | 0.23 – 0.27 | 0.08 – 0.7 | [0.15]Footnote f | 0.09 – 0.73 | |||
| Niacin | mcg | 250 | 345 – 5000 | 2800 – 4400 | 550 – 5000 | [720] | 900 – 5000 | |||
| Pantothenic acid | mcg | 300 | 300 – 1900 | 900 – 1500 | 300 – 1900 | [667 – 1083] | 450 – 1900 | |||
| Folic acid | mcg | 4 | 32 – 90 | 19 – 45 | 30 – 45 | [50]Footnote f | 32 – 91 | |||
| Biotin | mcg | 2 | 1.5 – 15 | 2.8 – 5.5 | 1.0 – 37 | [1.3] | 1.5 – 15 | |||
| Choline | mg | 12 | 7 – 50 | 11.1 – 25.5 | 7 – 23 | NS | 7.3 – 50 | |||
| Inositol | mg | NS | 4 – 48 | 25 – 74 | 4 – 44 | NS | 4 – 48 | |||
| Taurine | mg | NS | NS | 3.5 – 8.2 | 5 – 12 | NS | NS | |||
| Carnitine | mg | NS | NS | 2.2 – 2.6 | 2 – 5.9 | NS | NS | |||
| Calcium (Ca) | mg | 50 | 110 – 130 | 77 – 200 | 123 – 185 | [134 – 200] | 109 – 182 | |||
| Phosphorus (P) | mg | 25 | 55 – 80 | 46 – 127 | 82 – 109 | [65 – 98] | 55 – 127 | |||
| Ca:P | -- | 1.2 – 2 | NS | NS | 1.7 – 2.0:1 | NS | NS | |||
| Magnesium | mg | 6 | 7.5 – 13.6 | NS | 6.8 – 17 | [4.1 – 8.1] | 7.5 – 13.6 | |||
| Sodium | mg | 20 – 60 | 63 – 105 | 53 – 105 | 39 – 63 | [48 – 77] | 63 – 105 | |||
| Potassium | mg | 80 – 200 | 60 – 120 | 60 – 106 | 60 – 160 | [81 – 114] | 71 – 177 | |||
| Chloride | mg | 55 – 150 | 95 – 161 | 82 – 226 | 60 – 160 | [74 – 118] | 95 – 161 | |||
| Iron | mg | 0.15 | 1.8 – 2.7 | 1.538 – 3.636 | 1.7 – 3.0 | [1.7 – 2.5] | 1.8 – 2.7 | |||
| Zinc | mg | 0.5 | 1.0 – 1.8 | 0.769 – 2.727 | 1.1 – 1.5 | [0.42 – 0.67] | 1.3 – 2.3 | |||
| Copper | mcg | 60 | 90 – 120 | 92 – 136 | 100 – 250 | [58 – 101] | 90 – 210 | |||
| Manganese | mcg | 5 | 6.3 – 25 | 0.5 – 6.8 | 6.3 – 25 | [0.5 – 0.9] | 0.9 – 13.6 | |||
| Iodine | mcg | 5 | 10 – 50 | 7.7 – 54.5 | 6 – 35 | [26 – 53] | 9 – 50 | |||
| Selenium | mcg | NS | 4.5 – 9 | 1.0 – 4.1 | 1.8 – 5.0 | [2.6 – 3.9] | 4.5 – 9 | |||
| Chromium | mcg | NS | 0.027 – 1.12 | 0.08 – 2.05 | NS | 0.04 – 0.08 | 0.027 – 2.045 | |||
| Molybdenum | mcg | NS | 0.27 – 4.5 | 0.23 – 0.27 | NS | 0.16 – 0.32 | 0.27 – 4.5 | |||
| Fluoride | mcg | NS | 1.4 – 55 | NS | 0 – 25 | NS | 1.4 – 55 | |||
- NUCLEOTIDES
| Nutrient | FDR requirements | International expert recommendations | Nutritional profile | Specification | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Name | Unit | ESPGHAN (2010)Reference 1 | Tsang, et. al. (2005)Reference 2 >1000 g BW | LSRO (2002)Reference 3 | WHOReference 4 >1000 g BW (stabilization to term)Footnote a | Koletzko, et.al. (2014)Reference 5 | Min | Max | ||
| Total Nucleotides | mg | NS | NS | NS | NS | NS | NS | |||
| AMP | mg | NS | NS | 0.27 – 0.73 | NS | NS | NS | |||
| CMP | mg | NS | NS | 1.6 – 3.7 | NS | NS | NS | |||
| GMP | mg | NS | NS | 0.03 – 0.54 | NS | NS | NS | |||
| UMP | mg | NS | NS | 0.69 – 0.9 | NS | NS | NS | |||
Abbreviations
- ALA
- Alpha-linolenic acid
- AMP
- Adenosine monophosphate
- ARA
- Arachidonic acid
- BW
- Body weight
- Ca:P
- Calcium to phosphorous ratio
- CMP
- Cytosine monophosphate
- DHA
- Docosahexaenoic acid
- g
- gram
- GMP
- Guanosine monophosphate
- LA
- Linoleic acid
- LSRO
- Life Sciences Research Office
- Max
- Maximum
- mcg
- microgram
- mg
- milligram
- Min
- Minimum
- NS
- None specified
- PUFA
- Polyunsaturated fatty acids
- RE
- Retinol equivalents
- TE
- Tocopherol equivalents
- UMP
- Uridine monophosphate
- WHO
- World Health Organization.
References
- Reference 1
-
Agostoni C, Buonocore G, Carnielli VP, De Curtis M, Darmaun D, Decsi T, et al. Enteral nutrient supply for preterm infants: commentary from the European Society of Paediatric Gastroenterology, Hepatology, and Nutrition Committee on Nutrition. Journal of Pediatric Gastroenterology and Nutrition, 2010:50. 85-91.
- Reference 2
-
Tsang RC, Uauy R, Koletzko B, Zlotkin S. Nutrition of the preterm infant, Scientific basic and practical guidelines. Cincinnati, OH: Digital Educational Publishing Inc. 2005.
- Reference 3
-
Klein CJ. Nutrient requirements for preterm infant formulas. Journal of Nutrition, 2002;132:1395S-577S.
- Reference 4
-
Tudehope D. et al. Nutritional Needs of the Micropreterm Infant. Journal of Pediatrics, March 2013, Volume 162, Issue 3, Supplement, Pages S72–S80.
- Reference 5
-
Koletzko B, Poindexter B, Uauy R. Nutritional Care of Preterm Infants: Scientific Basis and Practical Guidelines. Karger, 2014:99-120.
Footnotes
- Footnote a
-
Calculated based on the average of the maximum and minimum energy recommendations for the indicated population; 120 kcal/kg/d for stabilization to term.
- Footnote b
-
3.6 – 4.1 g / 100 kcal for infants < 1 kg BW; 3.2 – 3.6 g/100 kcal for infants 1 - 1.8 kg BW
- Footnote c
-
Total fatty acids: 8% (minimum) and 25% (maximum)
- Footnote d
-
A minimum recommendation for ARA was not established. Maximum: 0.6% of total fatty acids with the further stipulation that the ARA:DHA is within the range of 1.5-2.0 : 1
- Footnote e
-
A minimum recommendation for DHA was not established. Maximum: 0.35% of total fatty acids with the further stipulation that the ARA:DHA is within the range of 1.5 – 2.0 : 1
- Footnote f
-
Recommendation given per day, independent of body weight and energy intake
- Footnote g
-
The ratio of vitamin E to PUFA (mg α-TE per g PUFA) should exceed 1.5 (mg α-TE/g PUFA)
- Footnote h
-
Recommendation per gram protein fed
Appendix 4.Summary of fluids and energy intakes recommended by international expert committees
| Expert committee | Fluid intake range (mL/kg/day) | Energy intake range (kcal/kg/day) | Weight of infant (g) |
|---|---|---|---|
| Koletzko (2014)Footnote 1 | 135-200 | 110-130 | 1000-1500 |
| ESPGHAN (2010)Footnote 2 | 135-200 | 110-135 | 1000-1800 |
| WHO (2006)Footnote 3 | N/A | 105-135 | >1000 |
| Tsang (2005)Footnote 4 and AAP (2014)Footnote 5 | 135-190 | 110-130* | 1000-1500 |
| LSRO (2002)Footnote 6 | 150 | 110-135 | 1000 |
Footnotes
* It is to be noted that for preterm infants with chronic lung disease, the recommended energy intake is increased to 140 kcal/kg/day, and for ELBW preterm infants, the maximum energy intake recommended is 150 kcal/kg/day. In special clinical situations intakes could reach 160 to 180 kcal/kg/day (AAP, 2014) |
|||
Appendix 5. Preterm human breast milk composition
NOTE: This data is recommended for use to fill the above template, and for standardisation of approach.
| Nutrients | Unit | Preterm Human Milk (per 100 kcal) |
Preterm Human Milk (per 100 mL) |
|---|---|---|---|
| MACRONUTRIENTS | |||
| ProteinReference 1 Footnote a | g | 2.1 | 1.5 |
| FatReference 1 Footnote a | g | 4.9 | 3.5 |
| - LA Reference 2 | g | 0.72 | 0.48 |
| - ALA Reference 2 | g | 0.045 | 0.03 |
| - LA:ALA | -- | 16:1 | 16:1 |
| - ARAReference 2 | g | 0.029 | 0.017 |
| - DHAReference 2 | g | 0.017 | 0.011 |
| Carbohydrate Reference 2 | g | 10.9 | 7.3 |
| VITAMINS AND MINERALS | |||
| Vitamin A Reference 2 | IU | 72 | 48 |
| Vitamin D Reference 2 | IU | 12 | 8 |
| Vitamin E Reference 2 | IU | 0.59 | 0.39 |
| Vitamin K Reference 2 | mcg | 3 | 2 |
| Vitamin C Reference 2 | mg | 6.6 | 4.4 |
| Thiamine Reference 2 | mcg | 13.35 | 8.9 |
| Riboflavin Reference 2 | mcg | 40.5 | 27 |
| Vitamin B6 Reference 2 | mcg | 9.3 | 6.2 |
| Vitamin B12 Reference 2 | mcg | 0.03 | 0.02 |
| Niacin Reference 2 | mcg | 315 | 210 |
| Pantothenic acid Reference 2 | mcg | 345 | 230 |
| Folic acid Reference 2 | mcg | 4.65 | 3.1 |
| Biotin Reference 2 | mcg | 0.81 | 0.54 |
| Choline Reference 3 | mg | 23.7 | 15.8 |
| Inositol Reference 4 | mg | 29.1 | 19.4 |
| VITAMINS AND MINERALS (continued) | |||
| Taurine Reference 2 | mg | 6 | 4 |
| Carnitine Reference 2 | mg | 1.05 | 0.7 |
| CalciumReference 1 Footnote a | mg | 35 | 25 |
| PhosphorusReference 1 Footnote a | mg | 21 | 15 |
| Ca:P | mg-mg | 1.67: 1 | 1.67: 1 |
| Magnesium Reference 2 | mg | 4.95 | 3.3 |
| Sodium Reference 2 | mg | 42 | 28 |
| Potassium Reference 2 | mg | 75 | 50 |
| Chloride Reference 2 | mg | 87 | 58 |
| Iron Reference 2 | mg | 0.135 | 0.09 |
| Zinc Reference 2 | mg | 0.56 | 0.37 |
| Copper Reference 2 | mcg | 57 | 38 |
| Manganese Reference 2 | mcg | 0.54 | 0.36 |
| Iodine Reference 2 | mcg | 26.7 | 17.8 |
| Selenium Reference 2 | mcg | 3.6 | 2.4 |
| ChromiumReference 5 Footnote b | mcg | 0.038 | 0.025 |
| Molybdenum Reference 5 | mcg | 0.3 | 0.2 |
| Fluoride Footnote c | mcg | NS d | NS |
| NUCLEOTIDES | |||
| Total Nucleotides | mg | NS | NS |
| AMP | mg | NS | NS |
| CMP | mg | NS | NS |
| GMP | mg | NS | NS |
| UMP | mg | NS | NS |
Abbreviations
- ALA
- Alpha-linolenic acid
- AMP
- Adenosine monophosphate
- BW
- Body weight
- ARA
- Arachidonic acid
- Ca:P
- Calcium to phosphorous ratio
- CMP
- Cytosine monophosphate
- DHA
- Docosahexaenoic acid
- g
- Gram
- GMP
- Guanosine monophosphate
- LA
- Linoleic acid
- mcg
- Microgram
- mg
- Milligram
- NS
- None specified
- UMP
- Uridine monophosphate
- WHO
- World Health Organization.
References
- Reference 1
-
Gidrewicz DA, Fenton TR. A systematic review and meta-analysis of the nutrient content of preterm and term breast milk. BMC Pediatr. 2014 Aug 30;14:216.
- Reference 2
-
Koletzko B, Poindexter B, Uauy R. Nutrtional Care of Preterm Infants. Scientific basis and practical guidelines. Basel, Switzerland: Karger AG; 2014.
- Reference 3
-
Maas C, Franz AR, Shunova A, Mathes M, Bleeker C, Poets CF, Schleicher E, Bernhard W. Choline and polyunsaturated fatty acids in preterm infants' maternal milk. Eur J Nutr. 2017 Jun;56(4):1733-1742.
- Reference 4
-
Moles L, Manzano S, Fernández L, Montilla A, Corzo N, Ares S, Rodríguez JM, Espinosa-Martos I. Bacteriological, biochemical, and immunological properties of colostrum and mature milk from mothers of extremely preterm infants. J Pediatr Gastroenterol Nutr. 2015 Jan;60(1):120-6.
- Reference 5
-
Klein CJ. Nutrient requirements for preterm infant formulas. J Nutr. 2002 Jun;132(6 Suppl 1):1395S-577S. [LSRO-2002]
Footnotes
For Reference #1, 71 kcal/100 mL; for all others, ~67 kcal/100 mL
- Footnote a
-
Protein, Fat, Calcium & Phosphorus values based on week 2 (from meta-analysis); All other values based on week 1-4
- Footnote b
-
Chromium values based on term human milk used by Life Sciences Research Office (LSRO 2002)
- Footnote c
-
NS due to wide range (likely due to variability of fluoride levels in water)