
Protocole d’essai clinique sur la croissance et la tolérance – Prématurés
Document d’orientation à l’intention des fabricants de préparations pour prématurés et de fortifiants pour lait humain
2021
Remerciements : Nous tenons à remercier Dre Tanis Fenton pour sa contribution à l'examen par les pairs de ce document d'orientation.
Table des matières
- 1.0 Introduction
- 2.0 Définitions
- 3.0 Protocole pour les essais cliniques sur la croissance et la tolérance pour soutenir le processus de soumission préalable à la mise en marché des préparations pour prématurés et des fortifiants pour lait humain.
- 3.1 Enregistrement
- 3.2 Titre de l’étude
- 3.3 Résumé
- 3.4 Introduction et renseignements généraux
- 3.5 Conception de l’étude
- 3.5.1 Description de la conception de l’étude
- 3.5.2 Objectifs de l’étude
- 3.5.3 Choix de la marge de non-infériorité ou de supériorité
- 3.5.4 Admissibilité des sujets
- 3.5.5 Taille de l’échantillon
- 3.5.6 Intervention
- 3.5.7 Durée de l’étude
- 3.5.8 Attribution du traitement
- 3.5.9 Insu
- 3.5.10 Collecte de données et résultats
- 3.5.11 Événements indésirables
- 3.5.12 Règles d’arrêt, interruptions, retraits et abandons
- 3.5.13 Procédures de contrôle de la qualité
- 3.5.14 Comité indépendant de surveillance des données
- 3.5.15 Violation du protocole
- 3.5.16 Modification du protocole
- 3.5.17 Analyses statistiques planifiées
- 3.6 Résultats de l’étude
- 3.7 Discussion et conclusions
- 4.0 Normes éthiques pour les chercheurs
- 5.0 Glossaire et références
- Appendice 1: Liste de contrôle CONSORT 2010 des renseignements à inclure dans les rapports d’un essai randomisé
- Appendice 2: Indicateurs biochimiques du bilan protéique et de l’état osseux chez les prématurés
- Appendice 3. Modèle des besoins nutritifs pour les préparations pour prématurés ou pour les fortifiants avec du lait humain (tels qu’ils sont donnés) par 100 kcal
- Appendice 4. Résumé des fluides et des apports nutritionnels recommandés par les comités d’experts internationaux
- Appendice 5. Composition du lait humain pour prématurés
1.0 Introduction
Toutes les préparations pour prématurés et les fortifiants pour lait humain doivent être sûrs et propres à la consommation humaine conformément à l’article 4.1 de la Loi sur les aliments et drogues. Ces aliments, destinés aux prématurés, sont réglementés en vertu du titre 25 de la partie B du Règlement sur les aliments et drogues (RAD) et doivent faire l’objet d’une évaluation préalable à la mise en marché par Santé Canada.
Les demandeurs doivent soumettre une notification préalable à la mise en marché pour toute nouvelle préparation pour prématurés ou tout nouveau fortifiant pour lait humain, ou pour un tel produit qui a fait l’objet d’un changement majeur à sa composition, à sa fabrication ou à son emballage. Les notifications doivent inclure la preuve établissant que la préparation pour prématurés ou le fortifiant pour lait humain est sûr et a une valeur nutritive adéquate pour favoriser une croissance et un développement acceptables chez les prématurés s’il est consommé conformément au mode d’emploi.
L’objectif de ce document est de fournir des directives aux fabricants concernant la conception appropriée d’une étude clinique de croissance et de tolérance. Santé Canada encourage les fabricants à consulter le Bureau des sciences de la nutrition sur le protocole d’essai clinique pour tout essai prévu, et à solliciter une réunion préalable à la demande d’approbation pour passer en revue la preuve requise pour une évaluation préalable à la mise en marché, comme les exigences peuvent varier.
2.0 Définitions
- Âge chronologique
Âge du nourrisson calculé en semaines à partir de la date de naissance, sans correction pour la prématurité (voir la figure 1).
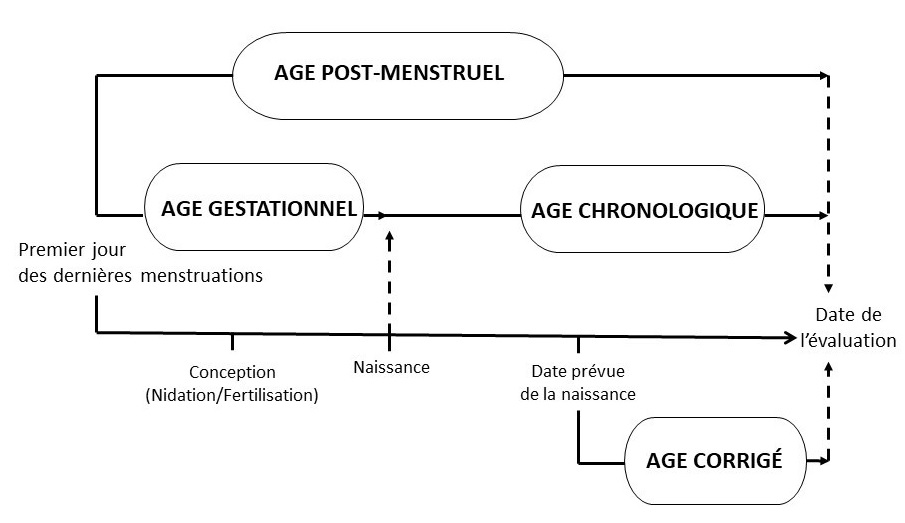
Figure 1 - Équivalent textuel
Chronologie montrant la relation entre l'âge post menstruel, l'âge gestationnel, l'âge chronologique et l'âge corrigé, tel que défini à la section 2.0.
- Âge corrigé [semaines ou mois] (c.-à-d. corrigé en fonction de la prématurité)
Âge chronologique, duquel est soustrait le nombre de semaines avant terme (avant 40 semaines de gestation); cette expression ne doit être utilisée que pour les enfants de moins de trois ans qui sont nés avant terme (voir la figure 1 ci-dessus (1)).
- Âge gestationnel
En obstétrique, âge du fœtus, calculé à partir de la date présumée du début des dernières menstruations normales (voir la figure 1) (2).
- Âge post-menstruel (APM)
Temps écoulé entre le premier jour des dernières menstruations et la naissance (âge gestationnel), plus le temps écoulé après la naissance (âge chronologique), en jours et semaines. Il s’agit du terme privilégié pour décrire l’âge d’un prématuré durant la période périnatale et l’hospitalisation. Après la période périnatale, « âge corrigé » est le terme privilégié (voir la figure 1 ci-dessus (1)).
- Alimentation entérale
Administration de tout aliment directement dans le tractus gastro-intestinal; cela comprend le gavage intragastrique, l’alimentation à la tasse ou au biberon et l’allaitement maternel (3).
- Alimentation orale
Administration de tout aliment dans la cavité buccale; cela comprend l’alimentation à la tasse, à la cuillère, à la seringue ou au biberon, l’expression directe et l’allaitement maternel, mais exclut le gavage par sonde gastrique (3).
- Changement majeur dans le cas d’un succédané de lait humain (article B.25.001 du RAD)
Signifie, en ce qui concerne un fortifiant pour lait humain ou un succédané de lait humain, tout changement apporté à un ingrédient ou à la quantité d’un ingrédient ou toute modification au traitement ou à l’emballage du fortifiant pour lait humain ou du succédané de lait humain, si l’expérience du fabricant ou la théorie généralement acceptée prévoit que ce changement aurait un effet indésirable sur les niveaux ou la disponibilité des éléments nutritifs ou sur l’innocuité microbiologique ou chimique du fortifiant pour lait humain ou du succédané de lait humain.
Exemples de changements majeurs apportés à une préparation pour prématurés ou à un fortifiant pour lait humain:
- changement important apporté au traitement ou à l’emballage de la préparation pour prématurés ou du fortifiant pour lait humain, ou modification de l’installation de fabrication ou ajout d’une nouvelle installation
- ajout d’une nouvelle source de macronutriments (protéines, lipides ou glucides)
- changement important de la teneur en protéines, en lipides ou en glucides
- ajout d’un nouvel ingrédient ou d’un aliment nouveau
- modification de la quantité ou des sources de vitamines ou de minéraux qui, selon l’expérience du fabricant ou la théorie généralement acceptée, aurait un effet indésirable sur :
- les niveaux ou la disponibilité des éléments nutritifs, ou
- l’innocuité microbiologique ou chimique
Pour de plus amples informations sur les données scientifiques nécessaires pour établir la valeur nutritive adéquate d’une nouvelle préparation pour prématurés ou d’un nouveau fortifiant pour lait humain, ou pour une telle préparation ou fortifiant ayant subi un changement important, veuillez consulter le Guide pour démontrer que la valeur nutritive est adéquate – Préparations pour prématurés et fortifiants pour lait humain.
- Fortifiant pour lait humain (article B.25.001 du RAD)
Désigne un aliment :
- dont la composition comprend une vitamine, un minéral nutritif ou un acide aminé ajouté,
- qui est étiqueté ou annoncé comme devant être ajouté au lait humain pour en augmenter la valeur nutritive afin de satisfaire aux besoins alimentaires particuliers d’un bébé qui manifeste un état physique ou physiologique particulier en raison d’une maladie, d’un désordre ou d’un état physique anormal.
- Lait humain enrichi
Mélange constitué d’un fortifiant pour lait humain et de lait humain pour prématurés.
- Lait humain enrichi d’essai
Signifie la formulation nouvelle ou modifiée du fortifiant combinée au lait humain pour prématurés.
- Lait humain enrichi témoin
Fortifiant approuvé au Canada avec du lait humain pour prématurés.
- Nutrition parentérale
Souvent appelé la nutrition parentérale totale, il s’agit du terme médical pour l’infusion d’une forme d’alimentation spécialisée par une veine (voie intraveineuse). L’objectif du traitement est de corriger ou de prévenir la malnutrition.
Remarque: Un fortifiant pour lait humain n’est jamais évalué seul sur le plan clinique. La combinaison de fortifiant et de lait humain est le produit final évalué. Dans ce document, le terme « lait humain enrichi » correspond à la combinaison du fortifiant et du lait humain.
- Période de croissance stable
Période qui commence lorsque le prématuré est stable sur le plan métabolique et clinique et qu’il prend du poids, et qui prend fin lorsque le prématuré atteint l’âge post-menstruel de 37 semaines (3) ou qu’il obtient son congé de l’hôpital.
- Période de transition
Période comprise entre la naissance et sept jours, durant laquelle les prématurés sont susceptibles d’être cliniquement et métaboliquement instables et de perdre du poids (3).
- Prématuré
Bébé né vivant avant la fin de la 37e semaine de grossesse. Il existe différentes sous-catégories de naissances prématurées, établies selon l’âge gestationnel (2) [veuillez consulter le tableau 1 ci-après].
|
Catégories |
Groupe |
Définition |
|---|---|---|
|
Âge gestationnel(2) |
Prématurité extrême/microprématurité | < 28 semaines |
| Grande prématurité | 28 à < 32 semaines | |
| Prématurité moyenne à tardive | 32 à < 37 semaines | |
|
Poids à la naissance(4) |
Faible poids à la naissance (FPN) | < 2500 g |
| Très faible poids à la naissance (TFPN) | < 1500 g | |
| Poids extrêmement faible à la naissance (PEFN) | < 1000 g | |
|
Poids selon l’âge(4) |
Petit pour l’âge gestationnel (PAG) | Poids à la naissance < 10e centile selon les courbes de croissance de référence utilisées pour les prématurés |
| Approprié pour l’âge gestationnel (AAG) | Poids à la naissance ≥ 10e et ≤ 90e centile selon les courbes de croissance de référence utilisées pour les prématurés | |
| Grand pour l’âge gestationnel (GAG) | Poids à la naissance > 90e centile selon les courbes de croissance de référence utilisées pour les prématurés |
- Prématurité
Catégorie globale de bébés nés à un âge gestationnel de moins de 37 semaines (2). La majorité des naissances prématurées surviennent entre 34 et 36 semaines de gestation, et représentent de 60 à 70 % de toutes les naissances prématurées; 15 % des naissances prématurées surviennent entre 28 et 31 semaines, et 5 % à moins de 28 semaines (5). Les prématurés peuvent aussi être subdivisés selon leur poids à la naissance, ou leur poids pour l’âge gestationnel, comme il est indiqué au tableau 1 ci-dessous. De plus, chaque catégorie est subdivisée selon différentes étapes de maturation fonctionnelle postnatales : transition, stabilisation et croissance établie.
Les prématurés et ceux ayant un faible poids à la naissance peuvent avoir de nombreuses difficultés physiologiques pendant leur adaptation au milieu extra-utérin et sont habituellement admis à une unité de soins intensifs néonatals (USIN) afin de stabiliser leur état et permettre un traitement médical spécialisé ainsi que des soins alimentaires. L’objectif des soins dispensés à l’USIN est d’évaluer et de surveiller les besoins de chaque nourrisson prématuré ou de faible poids à la naissance de façon individuelle et de donner le soutien approprié jusqu’à ce qu’il atteigne une maturité fonctionnelle (6).
- Préparation pour prématurés
Aliment présenté comme un remplacement partiel ou total du lait humain pour prématurés et qui est destiné à être consommé par des prématurés, de FPN, de TFPN ou de PEFN. Les préparations pour prématurés sont enrichies en calories (environ 80 kcal/100 mL contre 67 kcal/100 mL pour les préparations pour nourrissons à terme), en protéines et en minéraux, pour aider le prématuré à atteindre des taux d’accrétion des éléments nutritifs qui se rapprochent des taux intra-utérins. Les calories proviennent principalement des protéines, des lipides et des glucides. Les deux derniers macronutriments sont utilisés pour accroître la densité énergétique. L’équilibre entre les calories et les protéines est important pour favoriser la croissance de masse tissulaire maigre. Les préparations pour prématurés ont des teneurs en protéines, en sodium, en calcium, en phosphore, en zinc, en cuivre et en vitamines plus élevées que le lait humain non enrichi ou les préparations standard pour nourrissons.
- Retard de croissance intra-utérin (RCIU)
Incapacité de maintenir les taux de croissance intra-utérine prévus; le retard peut être dû à une insuffisance placentaire, une infection, une malnutrition, etc. (l’enfant peut ou non naître prématurément). Cela décrit un fœtus qui n’a pas atteint son potentiel de croissance à cause de facteurs génétiques ou environnementaux (7).
- Retard de croissance postnatal
Diminution du poids entre la naissance et la sortie de l’hôpital, supérieure à −2 scores Z selon les courbes de croissance pour prématurés de Fenton (8).
- Succédané de lait humain nouveau (préparations pour nourrissons) (article B.25.001 du RAD)
Désigne un succédané de lait humain :
- soit qui est fabriqué pour la première fois,
- soit qui est vendu au Canada pour la première fois,
- soit qu’une personne fabrique pour la première fois.
- Taux recommandé de prise de poids
Objectif de 15 à 20 g/kg/jour calculé après la phase de perte de poids postnatale, le plus souvent environ sept jours après la naissance, en s’appuyant soit sur le modèle exponentiel soit sur le modèle à deux points pour le calcul de la prise de poids moyenne (9, 10, 11, 12, 13). Cet objectif peut convenir à des nourrissons dont l’âge post-menstruel se situe entre 23 et 36 semaines (12, 13).
3.0 Protocole pour les essais cliniques sur la croissance et la tolérance pour soutenir le processus de soumission préalable à la mise en marché des préparations pour prématurés et des fortifiants pour lait humain
Les sections ci-dessous présentent toutes les exigences générales d’un protocole d’essai clinique pour une étude de croissance et de tolérance chez les prématurés cliniquement et métaboliquement stables, de faible poids ou de très faible poids à la naissance. Ces trois catégories de prématurés représentent la population étudiée à laquelle on fera référence dans le protocole comme des prématurés. Le rapport d’étude doit suivre le format précisé à l’appendice 1 (liste de contrôle CONSORT 2010) (14).
3.1 Enregistrement
L’enregistrement des essais cliniques doit répondre aux exigences suivantes:
- Le protocole doit être inscrit dans un registre d’essai clinique. On peut trouver des exemples de registres primaires d’essai sur le site Web de l’OMS (15).
- Le registre doit inclure tous les renseignements et critères précisés dans l’Ensemble des données sur l’enregistrement des essais cliniques de l’Organisation mondiale de la Santé (OMS) (16).
- Le numéro d’enregistrement et le nom du registre d’essai doivent être déclarés.
- L’enregistrement doit indiquer où on peut consulter le protocole complet de l’essai (17).
- Bien que des changements au protocole soient fortement découragés une fois l’essai en cours, l’enregistrement de l’essai clinique doit être mis à jour lorsque des modifications sont apportées ou s’il y a non-respect du protocole après le début de l’essai; les détails doivent être clairement décrits dans le rapport de l’essai (18).
3.2 Titre de l’étude
Le titre de l’étude doit préciser que l’étude est un essai de non-infériorité randomisé chez les prématurés (18,19).
Un essai de supériorité est approprié pour l’évaluation d’une nouvelle préparation pour prématurés ou d’un nouveau fortifiant pour lait humain proposé pour améliorer la croissance ou la tolérance alimentaire, ou pour tout autre effet sur la santé, par rapport aux résultats d’un produit concurrent témoin.
3.3 Résumé
Le protocole et le rapport d’étude doivent inclure un résumé qui décrit les objectifs, la conception et la méthode de l’essai, la collecte et l’analyse des données, ainsi que les résultats et les conclusions de l’étude (14).
3.4 Introduction et renseignements généraux
Les renseignements suivants doivent figurer dans l’introduction :
- nom et description des produits à l’étude, y compris leur forme physique (p.ex. poudre, liquide concentré, etc.) et confirmation que le produit témoin est approprié;
- justification pour l’essai clinique et une question d’étude clairement énoncée;
- description de la population de prématurés à étudier et ses similitudes avec la population cible;
- résumé des résultats des études cliniques et non cliniques pertinentes concernant les prématurés;
- résumés des risques et des bénéfices connus et potentiels, le cas échéant, pour les sujets humains, en particulier les prématurés.
3.5 Conception de l’étude
3.5.1 Description de la conception de l’étude
L’étude doit être conçue selon la norme d’excellence des essais cliniques : une étude prospective, randomisée, bien contrôlée, parallèle, monocentrique ou multicentrique et à double insu (20). Il faut inclure un diagramme schématique de la conception de l’essai, des procédures et des étapes (14).
3.5.2 Objectifs de l’étude
Les objectifs de l’étude devraient être d’évaluer les effets de la préparation pour prématurés d’essai ou d’un lait humain enrichi sur les mesures de croissance, de tolérance alimentaire et d’innocuité. L’étude doit également se faire par comparaison à un produit témoin concurrent approprié qui a déjà fait l’objet d’essais cliniques et dont l’innocuité et la valeur nutritive adéquate ont déjà été démontrées en fonction des courbes de croissance acceptables pour les prématurés (21).
3.5.2.1 Objectif principal
L’objectif principal de l’essai devrait être d’évaluer la valeur nutritive adéquateNote de bas de page 1 mesurée selon le taux de prise de poids sur une période minimale de 28 jours ou jusqu’à la sortie de l’hôpital (à compter de sept jours après la naissance ou une fois l’administration complète du produit débutée). Ce taux devrait être exprimé en g/kg/jour [calculé à l’aide du modèle exponentiel ou du modèle à deux points pour le calcul de la prise de poids moyenne] (9, 10, 11, 21, 22) chez les prématurés nourris avec la préparation ou le lait humain enrichi d’essai par rapport aux prématurés nourris avec un produit témoin concurrent (23).
Notes de bas de page
- Note de bas de page 1
-
Pour plus d’informations sur les données probantes requises par Santé Canada pour démontrer que les préparations pour nourrissons prématurés et de fortifiants pour lait humain offrent une valeur nutritive adéquate, veuillez consulter Guide pour démontrer que la valeur nutritive est adéquate - Préparations pour prématurés et fortifiants pour lait humain nourrissons.
L’objectif nutritionnel pour les prématurés est d’atteindre des taux de croissance et des taux d’accrétion des éléments nutritifs qui se rapprochent des taux atteints par les fœtus d’âge post-menstruel semblable in utero, tout en maintenant des concentrations normales d’éléments nutritifs dans le sang et autres tissus (24) et en évitant les complications qui peuvent découler de thérapies nutritionnelles agressives.
3.5.2.2 Objectifs secondaires
Les objectifs secondaires devraient être d’évaluer les mesures de croissance et d’innocuité suivantes et de faire les comparaisons subséquentes entre le groupe d’essai et le groupe témoin :
- temps nécessaire pour retrouver le poids à la naissance;
- taux d’augmentation de la taille et de la circonférence de la tête, exprimés en cm/semaine;
- mesures obtenues pour le poids, la taille et la circonférence de la tête enregistrées à tous les intervalles prédéterminés (toutes les semaines) pendant la durée de l’étude, pour les individus et les groupes, stratifiées selon le sexe, et la catégorie de poids à la naissance ou d’âge gestationnel et représentées sur la courbe de croissance pour prématurés de Fenton pour cette mesure de croissance et ce sexe (21);
- scores Z normalisés et changements aux scores Z (et leurs intervalles de confiance à 95 %) du groupe pour le poids et la taille ainsi que pour la circonférence de la tête selon l’âge ainsi que le poids selon la taille en fonction des courbes de croissance pour prématurés de Fenton (12, 21) à la naissance, au début de l’étude et à chaque visite dans le cadre de l’étude selon le sexe ou la catégorie de poids à la naissance ou d’âge gestationnel;
- temps avant une alimentation entérale complète, définie comme 150 ml/kg/jour (25);
- temps nécessaire pour atteindre un poids corporel de 1 800 à 2 000 g, ce qui est considéré comme une étape importante dans ce domaine de recherche (moment potentiel de la sortie de l’hôpital);
- volume moyen de la prise quotidienne de la préparation pour prématurés ou de lait humain enrichi (lait de la mère ou d’une donneuse), de calories, de protéines, de suppléments modulaires, de vitamines et de suppléments minéraux;
- incidence d’une perte de poids de plus de 2 scores Z selon les courbes de croissance pour prématurés de Fenton (21) à 36 semaines d’âge post-menstruel ou à la sortie de l’hôpital (8);
- durée de la nutrition parentérale totale (NPT) [jours];
- durée du séjour à l’hôpital (jours);
- proportion de nourrissons ayant au moins une période d’interruption de l’alimentation de ≥ 12 heures;
- évaluation à l’insu de la tolérance par le médecin (retenue de l’alimentation entérale, résidus gastriques, distension abdominale, coliques, vomissements, irritabilité, crampes, habitudes de sommeil, régurgitation, caractéristiques des selles, etc.);
- incidence des événements indésirables et des événements indésirables graves, tels que la mortalité, l’acidose métabolique, la défaillance ou l’insuffisance rénale, la détérioration de la fonction respiratoire, la septicémie, la rétinopathie de prématurité et l’entérocolite nécrosante néonatale (ENN), consignés sur un formulaire d’exposé de cas (FEC) par des professionnels de la santé;
- toutes les analyses biochimiques prévues au départ, au milieu et à la fin de l’étude, comme la mesure des taux de sérum-albumine, d’azote uréique sanguin (AUS), de créatinine, de calcium, de phosphore, de magnésium, d’hémoglobine, d’hématocrite, de fer sérique, de la capacité totale de fixation du fer, de la phosphatase alcaline sérique, des électrolytes sériques, des gaz sanguins et du pH (les analyses exigées seraient déterminées au cas par cas, en fonction de la nature de la préparation [voir l’appendice 2]).
3.5.3 Choix de la marge de non-infériorité ou de supériorité
Au moins une étude clinique dotée d’une puissance statistique suffisante dans la population cible doit être effectuée pour évaluer l’innocuité et déterminer que la valeur nutritive de la préparation pour prématurés d’essai ou du fortifiant pour lait humain est adéquate. La conception de l’essai clinique dépend de nombreux facteurs et des objectifs de l’étude.
Un essai de non-infériorité, avec une marge de non-infériorité (MNI) prédéfinie, est hautement recommandé quand l’objectif de l’étude est d’évaluer l’innocuité et de déterminer que la valeur nutritive est adéquate chez les prématurés nourris avec la préparation ou le lait humain enrichi d’essai par rapport à ceux nourris avec la préparation ou le fortifiant pour lait humain témoin commercial (19,20).
Il est recommandé d’utiliser un modèle de supériorité avec une marge de supériorité prédéfinie (26) si l’objectif de l’étude est de démontrer la supériorité d’une nouvelle préparation ou d’un nouveau lait humain enrichi d’essai par rapport à une préparation ou à un lait humain enrichi témoin approuvé.
Dans le cas où un essai de non-infériorité est initialement mené et que les résultats de l’essai révèlent des preuves suffisantes de supériorité, il est acceptable de remplacer l’objectif de non-infériorité par celui de supériorité. Pour ce faire, on peut calculer et évaluer la valeur p associée à un essai de supériorité dans l’analyse par intention de traiter (voir la section « Transition d’un essai de non-infériorité à un essai de supériorité » ci-après).
Essai de non-infériorité
Un essai de non-infériorité cherche à démontrer qu’une nouvelle préparation ou un nouveau lait humain enrichi d’essai n’est pas inférieur à la préparation pour prématurés ou au lait humain enrichi témoin. L’hypothèse de l’essai concernant la non-infériorité du taux de prise de poids chez les nourrissons recevant la préparation ou le lait humain enrichi d’essai par rapport à ceux recevant le produit témoin doit être clairement indiquée, en précisant la MNI, qui devrait être justifiée sur le plan clinique (19).
Il est très important de définir a priori la MNI. Pour déterminer la MNI, il faut s’appuyer sur une combinaison de considérations statistiques et de jugement clinique. La marge choisie pour un essai de non-infériorité ne doit pas être supérieure à la plus petite ampleur de l’effet du produit témoin. L’importance et la variabilité de l’ampleur de l’effet du produit témoin, calculées à partir d’essais historiques, devraient être prises en considération (26). De plus, la MNI sélectionnée devrait refléter les incertitudes dans les données probantes sur lesquelles le choix est basé, être suffisamment prudente et tenir compte de la variabilité (19, 26). Le protocole doit inclure un raisonnement ou de la littérature scientifique pour justifier le choix d’une telle marge.
Au cours de la dernière décennie, différents essais cliniques de la croissance et de la tolérance chez les prématurés ont sélectionné une différence moyenne qui variait de -1 à -4 g/kg/jour comme MNI cliniquement acceptable pour le taux de prise de poids (désignée par -δ), (27, 28,29). Dans l’ensemble, une MNI cliniquement acceptable de -1,6 g/kg/jour a été utilisée plus souvent que d’autres valeurs (29, 30, 31, 32). Ces essais cliniques n’ont toutefois pas fourni suffisamment de preuves ou de données historiques pour appuyer leur choix de MNI pour le taux de prise de poids. La méta-analyse et l’analyse systématique internes de Santé Canada (33) et les deux examens systématiques de Cochrane (34, 35), qui ont utilisé les données regroupées de différentes études cliniques de croissance et de tolérance chez les prématurés, ont démontré qu’une MNI de -1,1 g/kg/jour serait plus raisonnable et acceptable car elle est fondée sur des données cliniques. Cette MNI se situe dans la plage de la moitié de la différence moyenne dans le taux de prise de poids et de sa limite inférieure (33, 34), comme il est recommandé par la Food and Drug Administration (26), et est équivalente à l’écart-type (ET) de 0,25 recommandé pour le taux de prise de poids suggéré par Tyson (2002), où l’ET du taux de prise de poids chez les prématurés au cours d’une période d’étude de quatre semaines, commençant entre les 7 et 21 premiers jours de vie, est d’environ 4,2 g/kg/jour. L’ET de 4,2 g/kg/jour, qu’on trouve dans la méta-analyse et l’analyse systématique actuelles, est comparable à ce qui est déclaré (12, 27, 28). L’hypothèse selon laquelle le produit d’essai est non inférieur au produit témoin pourrait être établie si la limite inférieure de l’IC 95 % pour la différence du taux de prise de poids entre les deux groupes (produit d’essai - produit témoin) se situe au-dessus de la MNI de -1,1 g/kg/jour, comme on le voit dans la figure 2 (36, 37).
Différence moyenne du taux de gain de poids (T-C, exprimée en g/kg/jour)
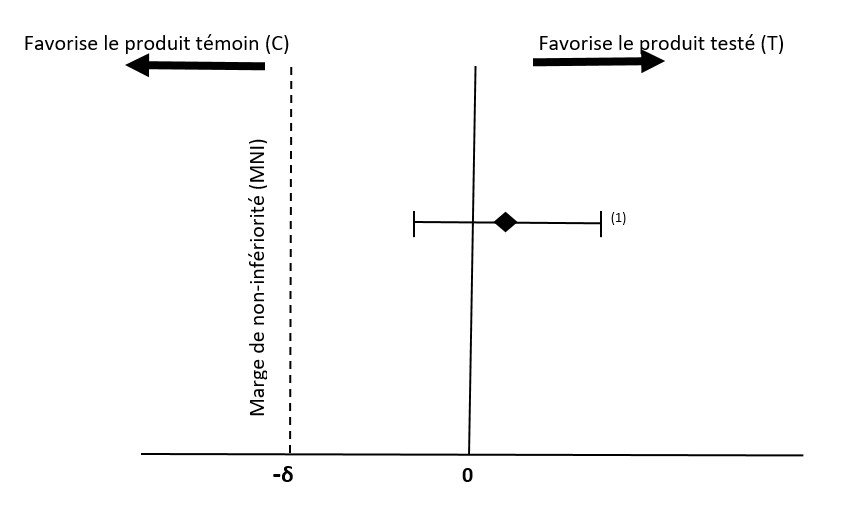
Figure 2 - Équivalent textuel
Illustre un résultat possible d'une étude de non-infériorité, montrant que le produit testé est non-inférieur au produit témoin.
Essai de supériorité
Un essai de supériorité cherche à démontrer la supériorité d’une nouvelle préparation ou d’un nouveau lait humain enrichi d’essai par rapport à une préparation ou à un lait humain enrichi témoin approuvé. L’hypothèse de l’essai concernant la supériorité des taux de croissance chez les nourrissons recevant la préparation ou le lait humain enrichi d’essai par rapport au produit témoin doit être clairement énoncée, et préciser la différence minimale cliniquement importante pour la croissance entre les nourrissons recevant la préparation ou le lait humain enrichi d’essai par rapport aux produits témoins, δ, ainsi que l’ampleur des risques d’erreurs de type I (α) et de type II (β) qui seraient acceptables pour l’essai. Pour qu’il y ait supériorité, plusieurs résultats doivent être évalués sans que rien n’indique des effets indésirables sur n’importe lequel des paramètres primaires ou secondaires cliniques importants.
Il arrive souvent qu’un essai de supériorité non significatif soit interprété à tort comme preuve qu’il n’y a pas de différence entre les deux traitements. En fait, l’essai de supériorité non significatif pourrait s’expliquer par une puissance statistique trop faible (par exemple, puissance de 80 %) pour détecter les différences entre le produit d’essai et le produit témoin. Il est donc très important de prédéfinir les valeurs utilisées pour α, β et δ afin de détecter la véritable différence, si elle existe. Les cliniciens, les chercheurs et les statisticiens devraient décider de ces valeurs a priori. Dans la plupart des cas, α, et β seraient précisés comme étant de 5 % et de 10 à 20 %, respectivement, mais dans des cas particuliers, des valeurs supérieures ou inférieures peuvent être utilisées pour les erreurs de type I et de type II. La valeur de δ doit être décidée selon des raisons cliniques a priori comme étant le taux de prise de poids le moins pertinent découlant de l’utilisation de la nouvelle préparation ou du nouveau lait humain enrichi d’essai, ainsi que de l’efficacité des produits témoins, et ce à quoi on pourrait raisonnablement s’attendre comme effet additif de la nouvelle préparation ou du nouveau lait humain enrichi d’essai. On peut s’appuyer sur des données préliminaires d’études pilotes ou des données d’observations historiques pour choisir la valeur de δ. Le protocole doit inclure un raisonnement ou de la littérature scientifique pour justifier le choix de telles valeurs pour α, β et δ.
Au minimum, la même marge (δ) de 1,1 g/kg/jour et la même différence de prise de poids de 4,2 g/kg/jour mentionnées dans l’essai de non-infériorité (voir la section ci-dessus) devraient être appliquées à l’essai de supériorité.
Transition d’un essai de non-infériorité à un essai de supériorité
Si les résultats d’une analyse par intention de traiter d’un essai de non-infériorité révèlent que le seuil de l’intervalle de confiance à 95 % pour l’effet du traitement ne se trouve non seulement entièrement au-dessus de -δ (figure 1) mais aussi au-dessus de zéro, cela constitue une preuve potentielle de supériorité. Dans ce cas, il faut évaluer la valeur p et le calcul de la puissance en lien avec un essai de supériorité pour déterminer s’il y a suffisamment de preuves pour rejeter l’hypothèse d’aucune différence en faveur de la supériorité, pourvu que les profils d’innocuité de la préparation et du lait humain enrichi d’essai et du produit témoin soient semblables.
Dans l’ensemble, on peut faire passer l’objectif d’un essai de non-infériorité à un objectif de supériorité si les exigences suivantes sont respectées :
- l’essai a été bien conçu et réalisé conformément aux exigences strictes d’un essai de non-infériorité et respecte les lignes directrices CONSORT;
- les valeurs p réelles de supériorité sont présentées pour permettre l’évaluation indépendante du poids de la preuve;
- l’importance la plus importante a été accordée à l’analyse selon le principe de l’intention de traiter dans l’essai de supériorité (38);
- aucune augmentation clinique ou statistique des événements indésirables n’est relevée.
3.5.4 Admissibilité des sujets
Les sujets de l’étude doivent être des nourrisons prématurés, FPN ou TFPN, de moins de 37 semaines d’âge gestationnel qui sont hospitalisés dans des USIN. Pour des raisons éthiques, pour un essai clinique de préparation pour prématurés, les mères doivent déjà avoir décidé de ne pas allaiter leur enfant. Les méthodes de recrutement, les milieux cliniques et les lieux géographiques où les critères d’admissibilité seront recueillis doivent être déclarés (39). Les dates définissant les périodes de recrutement et de suivi, ainsi que des renseignements sur la façon dont on prévoit faire le suivi de ces sujets doivent être fournis. Il faut inclure des prématurés de sexe masculin et féminin de manière égale dans l’étude, à des chiffres qui respectent le calcul de la puissance.
Les critères d’inclusion et d’exclusion ci-dessous doivent être appliqués.
Critères d’inclusion
- Prématurés, de faible poids ou de très faible poids à la naissance qui sont cliniquement et métaboliquement stables;
- Prématurés uniques, jumeaux ou triplés de < 37 semaines de gestation (les deux jumeaux ou les trois triplés n’ont pas à se qualifier pour l’étude ou à être randomisés dans celle-ci);
- Poids à la naissance ≤ 1 800 g;
- Poids approprié pour l’âge gestationnel (voir le tableau ci-dessus pour la définition);
- Hospitalisation prévue pendant au moins la durée de l’étude après l’inscription à l’étude;
- Intention de la mère de fournir du lait humain pendant l’étude dans le cas de l’évaluation d’un nouveau lait humain enrichi (permettre le lait de donneuses, au besoin);
- Nourrisson âgé de 21 jours ou moins au moment de l’inscription;
- Alimentation surtout à base de préparation ou de lait humain enrichi avant le début de l’étude;
- Tolérance de l’alimentation entérale, y compris l’alimentation trophique;
- Aucune utilisation de stéroïdes à la randomisation et aucune intention de commencer des stéroïdes pendant l’essai;
- Approbation éthique par un comité d’éthique pour la recherche avec des êtres humains;
- Formulaire de consentement signé obtenu d’un parent ou d’un tuteur légal;
- Alimentation entérale ayant commencé dans les 7 à 14 premiers jours de vie.
Critères d’exclusion
- Antécédents d’une maladie, d’un problème de santé ou d’une malformation congénitale sous-jacent, qui, de l’avis du chercheur, seraient susceptibles de nuire à la croissance et au développement normaux ou à l’évaluation du participant;
- Sujet ayant subi une grosse chirurgie qui a nécessité une anesthésie générale avant la randomisation;
- Sujet petit pour l’âge gestationnel, c’est-à-dire d’une taille inférieure au 10ecentile sur une courbe de croissance pour prématurés de Fenton propre au sexe;
- Restriction liquidienne < 120 ml/kg/jour;
- Hémorragie périventriculaire ou intraventriculaire;
- ENN supérieure au stade II, septicémie confirmée, défaillance ou insuffisance rénale, incapacité maternelle (y compris la consommation de cocaïne ou d’alcool par la mère pendant la grossesse ou au moment de l’inscription à l’essai), séropositivité VIH de la mère ou du nourrisson, asphyxie menant à des dommages neurologiques graves et permanents, convulsions, ou infection systémique non contrôlée au moment de l’admission à l’essai;
- Participation à une autre étude clinique qui n’a pas été approuvé par le promoteur;
- Antécédents maternels ayant des effets néfastes connus sur le fœtus ou le nouveau-né, comme le diabète, la tuberculose progressive, une infection périnatale ou la toxicomanie;
- Indice d’Apgar évalué à cinq minutes < 4 [apparence, pouls, grimace (expression déformée) activité et respiration];
- Probabilité raisonnable d’un transfert à une autre USIN où on n’a pas obtenu une approbation relative à l’éthique de la recherche avec des êtres humains;
3.5.5 Taille de l’échantillon
Le calcul de la taille de l’échantillon devrait être basé sur le résultat principal, avec des méthodes ou des critères particulièrement conçus pour un essai de non-infériorité ou de supériorité. Différents facteurs sont pris en compte pour déterminer la bonne taille d’échantillon, y compris le choix d’une MNI ou de supériorité et la puissance statistique.
Le choix d’une MNI, qui repose sur une différence cliniquement importante dans le taux de prise de poids au cours de la durée de l’étude, devrait être déterminé selon les preuves statistiques (par exemple, résultats de la méta-analyse et de l’analyse systématique d’études récentes de croissance et de tolérance chez les prématurés) et le jugement clinique (26). L’écart-type du taux de prise de poids, tenant compte notamment de la variabilité dans le sexe, du poids à la naissance et de l’âge gestationnel, devrait être déterminé en fonction de données historiques (par exemple, données concernant le taux de prise de poids chez les prématurés découlant de plusieurs essais cliniques sur la croissance menés auprès de prématurés).
L’échantillon doit être suffisamment grand pour avoir une forte probabilité (puissance) de détecter une différence statistiquement significative correspondant au moins à la marge, si une telle différence existe entre les groupes, en utilisant l’analyse prévue.
Comme il est expliqué ci-dessus, une MNI de -1,1 g/kg/jour et une différence minimale cliniquement importante de 1,1 g/kg/jour du taux de prise de poids pour les prématurés, au cours d’un intervalle d’étude de 28 à 31 jours, commençant entre les premiers 7 à 21 jours de vie, sont considérées comme étant les différences minimales cliniquement acceptables pour le taux de prise de poids entre le groupe d’essai et le groupe témoin. Des études récentes (12, 28, 33) suggèrent également que l’écart-type du taux de prise de poids pour une période d’étude de quatre semaines, commençant dans les 21 premiers jours de vie, est d’environ 4,2 g/kg/jour.
Par conséquent, dans le cas d’un essai de non-infériorité, il est recommandé que la taille de l’échantillon soit calculée pour déterminer si la préparation pour prématurés ou le lait humain enrichi d’essai est inférieur au produit témoin, selon une MNI de -1,1 g/jour et un écart-type commun de 4,2 g/kg/jour. Plus précisément la valeur α serait précisée entre 0,025 et 0,05, et la valeur β, entre 10 % et 20 %; le taux d’abandon prévu se situerait entre 30 et 35 %, respectivement.
Pour un essai de supériorité de croissance et de tolérance chez les prématurés, la taille de l’échantillon devrait être calculée pour évaluer si la préparation pour prématurés ou le lait humain enrichi expérimental est supérieur ou non au produit témoin approuvé concurrent avec suffisamment de puissance pour détecter la différence minimale cliniquement importante de 1,1 g/kg/jour, supposant que l’écart-type commun est de 4,2 g/kg/jour. Les valeurs α et β ainsi que le taux d’abandon prévu seraient souvent précisés comme pour l’essai de non-infériorité.
3.5.6 Intervention
Les détails précis de l’intervention prévue pour chaque groupe doivent être déclarés. Il est recommandé que les sujets soient affectés aux groupes suivants selon un rapport équilibré.
- Produit d’essai
Le produit d’essai doit être celui proposé pour le marché canadien, c’est-à-dire la même composition, le même profil nutritionnel et le même processus de fabrication, et il doit être conforme aux exigences nutritives du RAD (concernant les préparations pour prématurés). Il doit être conforme ou légèrement supérieur aux recommandations concernant les nutriments des experts internationaux pour les prématurés (voir l’appendice 3 ci-jointe).
- Produit témoin
Un produit témoin concurrent doit être inclus dans l’étude. Il est recommandé que le produit témoin soit une préparation pour prématurés commerciale canadienne ou un fortifiant pour lait humain commercial canadien + lait humain pour prématurés.
Le profil nutritionnel de la combinaison du fortifiant pour lait humain + lait humain pour prématurés pour chaque groupe devrait être inclus dans le rapport d’étude et présenté dans la soumission préalable à la mise en marché. On recommande que le profil nutritionnel repose sur l’analyse de deux ou trois échantillons de ces combinaisons au moment de l’alimentation entérale complète de lait humain enrichi.
Si la mère ne produit pas suffisamment de lait, du lait provenant de donneuses (mères de nourrissons à terme) peut être utilisé en association avec un fortifiant pour lait humain; toutefois le lait de la mère du prématuré est recommandé (40). Voir les appendices 4 et 5 pour la composition du lait humain pour prématurés et les différents volumes et teneurs énergétiques utilisés par les experts pour formuler leurs recommandations nutritionnelles.
Dans le cas d’une préparation pour prématurés ou d’un fortifiant pour lait humain auquel on a ajouté un nouvel ingrédient, le produit d’essai et le produit témoin doivent être identiques à l’exception du nouvel ingrédient.
- Comparaison
Le protocole doit relever et décrire toutes les différences dans la composition et les profils nutritionnels, le cas échéant, y compris la densité énergétique et la source de protéines du produit d’essai et du produit témoin.
3.5.7 Durée de l’étude
Pendant au moins quatre semaines ou jusqu’à leur sortie de l’hôpital, les sujets doivent exclusivement (c.-à-d. plus de 80 % de leur consommation quotidienne) recevoir la préparation pour prématurés ou le lait humain fortifié qui leur est attribué. Un suivi à plus long terme (c.-à-d. jusqu’à 36 à 40 semaines d’âge corrigé) est fortement recommandé.
3.5.8 Attribution du traitement
La répartition des participants entre le groupe d’essai et le groupe témoin doit se faire de façon aléatoire et dissimulée, et dans le respect des lignes directrices CONSORT (14, 19). La randomisation réduit les biais de sélection en tenant compte des facteurs de confusion connus et inconnus dans la population d’étude, quand elle est bien effectuée et est dissimulée. Le processus de randomisation comprend les aspects suivants (14, 19, 39) :
- Génération de séquence
Décrire la méthode utilisée pour générer la séquence de répartition aléatoire. Les méthodes adéquates comprennent l’utilisation d’un tableau de nombres aléatoires ou d’un générateur de nombres aléatoires informatisé (19). La randomisation des sujets devrait être stratifiée selon la catégorie de poids de naissance, le lieu (dans un essai multicentrique) et le sexe, comme les garçons n’ont pas le même taux de croissance que les filles.
- Dissimulation de la répartition
La séquence de répartition aléatoire doit être dissimulée jusqu’à ce que le recrutement soit terminé et irrévocable, et que les interventions ont été attribuées. Par exemple, la répartition du traitement pourrait être dissimulée par un outil de randomisation ou de répartition informatisé ou en dissimulant le traitement assigné dans des contenants opaques, scellés et numérotés dans l’ordre, ou dans des enveloppes opaques scellées et agrafées (39).
On recommande de bons mécanismes de dissimulation de la répartition des traitements, qui intègrent une participation externe (deux techniques courantes sont l’utilisation d’une pharmacie ou d’un système de randomisation par téléphone central) (39).
Mise en œuvre de la randomisation
Le protocole doit inclure des renseignements détaillés sur tout le personnel chargé de générer la séquence aléatoire, de recruter des participants et de répartir les participants entre les différents groupes de l’étude (39). Toute déviation par rapport à la répartition randomisée doit être décrite dans le rapport d’étude.
3.5.9 Insu
Le traitement utilisé ne doit pas être révélé aux participants à l’étude (parents ou personnes soignantes), aux chercheurs, aux évaluateurs des résultats ou aux autres employés de l’étude. Les produits d’essai et les produits témoins doivent être d’apparence identique; par exemple, dans le cas de préparations, l’emballage, la texture, le goût et l’odeur doivent être les mêmes (39). Des renseignements détaillés doivent être fournis concernant la façon dont les produits sont étiquetés (par exemple, par code de sujet individuel), les personnes qui ont accès aux codes de produits, les circonstances prédéfinies dans lesquelles l’insu pourrait être levé, le cas échéant, et les membres de l’équipe de chercheurs pour qui l’insu serait levé en cas d’un tel besoin. De plus, il faut maintenir l’insu jusqu’à la fin de l’étude. Les évaluateurs des résultats doivent être maintenus à l’insu en tout temps.
3.5.10 Collecte de données et résultats
3.5.10.1 Calendrier de collecte de données
Il faut décrire le calendrier de collecte de données. Les sujets du groupe d’essai et du groupe témoin doivent être inscrits et suivis en parallèle. Les protocoles standard d’alimentation clinique des USIN doivent être fournis.
Les données de mesure doivent être prélevées en respectant les procédures normalisées, avec des instruments étalonnés. La formation du personnel de l’étude, ainsi que les procédures d’assurance de la qualité, doivent être en place pour veiller à ce que les mesures soient aussi précises et exactes que possible pour tous les participants, sites d’étude et moment d’enregistrement des données. Si les données recueillies ne sont pas complètes, la portée et l’incidence des données manquantes doivent être décrites dans le rapport d’étude, et l’approche adoptée pour tenir compte des données manquantes doit être déclarée.
3.5.10.2 Collecte des renseignements démographiques de base
Tous les nourrissons inclus dans l’étude clinique doivent être bien caractérisés, surtout en ce qui concerne les facteurs qui pourraient influer sur les résultats prévus. Afin de permettre une évaluation scientifique complète de l’étude, les renseignements suivants doivent être fournis :
Données sur le nourrisson
- Sexe du nourrisson;
- Méthode d’accouchement (accouchement vaginal ou césarienne);
- Naissance unique, jumeaux ou triplés;
- Âge gestationnel à la naissance et au début de l’essai;
- Poids à la naissance par gestation (approprié pour l’âge gestationnel, ou autre);
- Données d’anthropométrie au début de l’essai, y compris le poids corporel, la taille et la circonférence de la tête en valeurs absolues et en scores Z, en consignant la norme de croissance utilisée pour calculer les scores Z. Consulter les liens suivants pour des explications sur la façon de mesurer le poids, la taille et la circonférence de la tête des nourrissons : http://depts.washington.edu/growing/Assess/Anthro.htm;
- Indice d’Apgar évalué à cinq minutes [apparence, pouls, grimace (expression déformée), activité et respiration];
- Âge chronologique au recrutement, à la randomisation et au début de chaque intervention et à chaque point d’évaluation;
- Race ou ethnicité;
- Utilisation simultanée de médicaments (caféine, stéroïdes, etc.) ;
- Conditions cliniques sous-jacentes, gravité et sévérité;
- Stéroïdes prénataux
- Âge en jours quand l’alimentation entérale a commencé;
- Âge au début de l’enrichissement;
- Courbes de croissance utilisées.
Données de la mère
- Âge de la mère, éducation et indice de masse corporelle (IMC);
- Antécédents de tabagisme pendant la grossesse ou à la randomisation;
- Alcoolisme ou toxicomanie de la mère pendant la grossesse ou à la randomisation;
- Parité de la mère;
- Antécédents maternels ayant des effets néfastes connus sur le fœtus ou le nouveau-né, comme le diabète, la tuberculose progressive ou une infection périnatale.
Données du père
- Âge du père et éducation;
- Taille et poids des parents, mesurés si possible;
- Statut socioéconomique.
3.5.10.3 Collecte de données sur la prise alimentaire
Des systèmes appropriés doivent être en place pour évaluer la conformité à la consommation quotidienne actuelle de la préparation ou du lait humain enrichi attribué pendant l’essai. Ces données sont importantes pour l’interprétation des résultats de l’essai.
3.5.10.4 Mesure des résultats
Les paramètres des résultats suivants doivent être évalués à des moments prédéterminés au cours de la durée de l’étude. Toutes les méthodes utilisées pour améliorer la qualité des mesures (observations multiples, formation des évaluateurs, etc.) doivent être déclarées.
Résultat principal
- Taux de prise de poids au cours de l’étude exprimé en g/kg/jour, calculé à l’aide du modèle exponentiel ou du modèle à deux points pour le calcul de la prise de poids moyenne (9, 10, 11, 22) entre le jour 0 de l’étude et sa fin.
Résultats secondaires
Les résultats suivants sur la croissance et l’innocuité devraient faire l’objet de comparaisons entre le groupe d’essai et le groupe témoin :
- taux hebdomadaires de gains de taille (cm/semaine) et de circonférence de la tête (cm/semaine) entre le jour 0 de l’étude et sa fin;
- poids, taille et circonférence de la tête mesurés au début de l’étude puis à chaque visite pour chaque nourrisson et chaque groupe stratifié selon le sexe, ou la catégorie de poids à la naissance ou d’âge gestationnel, représentés sur les courbes de croissance pour prématurés de Fenton (21);
- scores Z calculés pour le groupe et changements aux scores Z et à leurs intervalles de confiance à 95 % pour le poids, la taille et la circonférence de la tête en fonction de l’âge, et le poids en fonction de la taille, à la naissance, au début de l’étude, à chaque visite de l’étude et à la sortie de l’hôpital au moyen des courbes de croissance pour prématurés de Fenton (21) selon le sexe, et les catégories de poids à la naissance et d’âge gestationnel;
- temps nécessaire pour retrouver le poids à la naissance;
- temps nécessaire pour atteindre un poids corporel de 1 800 à 2 000 g, ce qui est considéré comme une étape importante dans ce domaine de recherche (moment potentiel de la sortie de l’hôpital, au cas par cas);
- temps pour commencer l’enrichissement;
- temps pour atteindre l’alimentation entérale complète, c’est-à-dire 150 ml/kg/jour;
- incidence d’une perte de poids de plus de 2 scores Z selon les courbes de croissance de prématurés pour Fenton (21) à 36 semaines ou à la sortie de l’hôpital (8);
- nombre de nourrissons petits pour l’âge gestationnel à leur sortie de l’hôpital, c’est-à-dire ayant un poids corporel inférieur au 10ecentile selon leur âge gestationnel donné par rapport aux courbes de croissance pour prématurés de Fenton (20);
- durée de la nutrition parentérale totale (NPT) [jours];
- durée de la nutrition entérale;
- durée des produits de l’étude;
- durée du séjour à l’hôpital (jours);
- proportion de nourrissons ayant au moins une période d’interruption de l’alimentation de ≥ 12 heures;
- incidence et enregistrements quotidiens des événements indésirables et des événements indésirables graves (mortalité, défaillance ou insuffisance rénale, septicémie, ENN, etc.) déclarés, vérifiés et évalués par des professionnels de la santé qui ne sont pas au su du traitement attribué;
- volume moyen de la prise quotidienne de la préparation pour prématurés ou du lait humain enrichi (lait de la mère ou d’une donneuse), de calories, de protéines, de suppléments modulaires, ainsi que de vitamines et de suppléments minéraux;
- registres quotidiens des données sur la tolérance et de l’évaluation à l’insu par des médecins (retenue de l’alimentation entérale, résidus gastriques, distension abdominale, vomissements, coliques, irritabilité, crampes, habitudes de sommeil, régurgitation, caractéristiques des selles, hospitalisation, etc.);
- fréquence et sévérité des événements indésirables et des événements indésirables graves et nombre total de participants chez qui ces événements ont été signalés;
- toutes les analyses biochimiques prévues au départ, au milieu et à la fin de l’étude, par exemple, créatinine, préalbumine, azote uréique sanguin, phosphatase alcaline sérique, électrolytes sériques, calcium, phosphore, magnésium, hémoglobine, hématocrite, fer sérique, capacité totale de fixation du fer, gaz sanguins, pH sanguin, pH urinaire, etc. Les analyses exigées doivent être déterminées au cas par cas, en fonction de la nature de la préparation;
- jours de ventilation en pression positive intermittente (VPPI);
- jours de ventilation en pression positive continue (VPPC);
- jours de ventilation mécanique;
- jours avec supplément d’oxygène ou probiotiques, noms des probiotiques fournis et moments d’administration;
- proportion de prise du lait de sa mère ou d’une donneuse;
- nombre de nourrissons avec une ENN, selon les critères de Bell > stade II;
- nombre de cas présumés de septicémie traités par antibiotiques;
- nombre de cas avérés de septicémie traités par antibiotiques;
- nombre de nourrissons ayant eu leur congé de l’hôpital ou ayant été transférés à une autre installation, et leur âge;
- nombre de cas de non-conformité ou de retrait.
Remarques : Pour les études de suivi à plus long terme, les Normes de croissance de l’enfant de l’Organisation mondiale de la Santé (2006) devraient être utilisées pour évaluer la croissance sur une durée d’étude supérieure à 50 semaines d’âge post-menstruel.
Surveillance après la mise sur le marché
Un plan de surveillance après la mise en marché peut être exigé pour étayer l’innocuité à long terme (41). La surveillance après la mise sur le marché consiste en :
- un volet de surveillance, qui prévoit des mesures pour surveiller les effets indésirables après la mise en marché du produit;
- un volet de suivi, qui met l’accent sur les effets retardés ou à long terme qui pourraient se produire après la période d’utilisation maximale du produit. La durée du suivi doit être déterminée au cas par cas;
- un rapport écrit semestriel ou annuel du comité médical et scientifique du demandeur, résumant les résultats de sa revue de la littérature, notamment ceux portant sur l’innocuité et le suivi à long terme de tout nouvel ingrédient ajouté (par exemple, lutéine);
- évaluation des résultats de croissance et de développement (au cas par cas).
3.5.11 Événements indésirables
Des renseignements précis et clairs sur les événements indésirables et les effets secondaires (non attendus) observés durant l’étude, ou durant une période de suivi prédéterminée, doivent être soigneusement consignés et déclarés, que ces événements ou effets soient ou non immédiatement associés au traitement à l’étude (42).
Le protocole pour établir avec certitude les événements indésirables et les effets secondaires possibles doit être défini a priori et déclaré. L’information à fournir comprend la durée et la fréquence des activités de surveillance, et une définition de cas valide et reconnue pour les événements indésirables courants ou prévus avec des critères normalisés, des listes de contrôle ou bien des échelles ou des scores de diagnostic valides pour les diagnostics, le cas échéant. Il faut également indiquer le nom des personnes ayant détecté ou confirmé l’événement indésirable (chercheur, dossiers médicaux, professionnel de la santé, personnel clinique, promoteur). Le cas échéant, il faut consigner la méthode pour traiter et compter les événements récurrents. Toutes les lignes directrices sur l’arrêt de l’essai en lien avec les événements indésirables doivent être définies et décrites.
En cas d’événements indésirables, les renseignements suivants doivent être consignés dans les dossiers médicaux et le rapport d’étude, et être confirmés par des professionnels de la santé au besoin :
- numéro du sujet, date et durée de l’événement;
- description de l’événement;
- sévérité (légère, modérée, sévère);
- gravité et fréquence;
- attribution potentielle à l’alimentation de l’étude;
- mesures prises et détails de tout médicament utilisé;
- retrait du participant après l’événement indésirable.
3.5.12 Règles d’arrêt, interruptions, retraits et abandons
Le protocole doit fournir des détails sur la procédure de surveillance des événements indésirables graves et indiquer les règles d’arrêt, ainsi que les critères d’interruption et ceux de retrait pour les sujets individuels, soit pour une partie de l’étude, soit pour toute sa durée, ainsi que le plan de suivi des sujets qui ont fait l’objet d’un retrait ou qui ont abandonné l’étude (42).
Des mécanismes doivent être en place pour réduire autant que possible les retraits de l’étude, pour des raisons autres que des événements indésirables. Le chercheur principal doit faire des efforts raisonnables pour déterminer les raisons de l’abandon dans tous les cas où un participant se retire de l’étude avant sa fin. Les détails suivants sur les sujets qui ne se sont pas conformés aux exigences, ou qui se retirent de l’essai ou l’abandonnent doivent être bien saisis et déclarés :
- description détaillée des participants qui ne se sont pas conformés aux exigences, ou qui se sont retirés de l’étude ou qui l’ont abandonné;
- description des raisons du retrait ou de l’abandon de l’étude;
- date et âge au moment de l’arrêt de la participation selon le protocole;
- détails de l’alimentation (alimentation parentérale, préparation pour prématurés, lait humain, lait humain enrichi et suppléments modulaires) avant l’abandon ou le retrait;
- durée de participation du sujet à l’étude avant l’abandon;
- raisons de la non-conformité;
- âge postnatal au retrait ou à l’abandon de l’étude.
3.5.13 Procédures de contrôle de la qualité
Le protocole doit décrire les procédures de contrôle de la qualité avec des procédures opérationnelles normalisées (PON) écrites pour garantir que l’essai est réalisé et que les données sont produites, consignées (enregistrées) et déclarées conformément au protocole, aux bonnes pratiques cliniques (BPC) et aux exigences réglementaires pertinentes (43). Les PON garantissent également le bon déroulement du recrutement de participants à l’essai afin de minimiser les sources potentielles de « biais de sélection » et veillent à ce que toutes les mesures de résultat au cours de l’étude sont exactes, reproductibles, valables et fiables.
3.5.14 Comité indépendant de surveillance des données
Le promoteur doit créer un comité indépendant de surveillance des données pour évaluer périodiquement les progrès de l’essai clinique, les données sur l’innocuité et les paramètres d’efficacité, et pour recommander au promoteur s’il faut continuer l’essai, le modifier ou y mettre fin. Le comité doit avoir des PON écrites et tenir des dossiers de toutes ses réunions, y compris celles concernant les résultats intermédiaires. Les membres du comité doivent inclure un biostatisticien ayant des connaissances sur la conception d’essais cliniques et sur les analyses, des scientifiques ayant de l’expérience liée aux études cliniques et possédant des connaissances dans le domaine, et un néonatologiste (18, 43).
3.5.15 Violation du protocole
Les violations de protocole doivent être déclarées dans le rapport de l’étude et le registre doit être mis à jour. Le rapport de l’étude doit consigner l’évaluation de l’ampleur de ces violations et la mesure dans laquelle elles peuvent influer sur l’interprétation des résultats (18, 44).
3.5.16 Modification du protocole
Les dérogations au protocole ou toutes les modifications importantes requises pendant l’étude doivent figurer dans le rapport d’étude et le protocole d’étude, et le registre doit être mis à jour, en notant précisément qu’un changement a été apporté. La justification pour toute modification importante et son influence sur l’interprétation des résultats doivent être évaluées et déclarées dans le rapport et le protocole d’étude; l’enregistrement de l’étude doit être mis à jour (18).
3.5.17 Analyses statistiques planifiées
Dans l’ensemble, l’analyse statistique et les méthodes utilisées doivent être conformes aux principes scientifiques généralement acceptés (45).
Le protocole doit clairement indiquer les hypothèses d’essai (par exemple, l’hypothèse d’un essai de non-infériorité ou de supériorité) ou les effets du traitement, qui doivent être estimés afin de satisfaire à l’objectif principal et aux objectifs secondaires de l’essai. Les méthodes statistiques utilisées pour accomplir ces tâches doivent être clairement décrites pour l’analyse principale ainsi que pour les analyses secondaires, et le modèle statistique sous-jacent doit être précisé a priori. Une description doit être donnée de toutes les intentions d’utiliser les données de référence pour améliorer la précision ou d’ajuster les estimations pour les différences de départ potentielles, par exemple au moyen d’une analyse de covariance.
La taille de l’échantillon doit être basée sur la détection d’une ampleur d’effet pour le résultat principal, qui est significatif tant sur le plan statistique que sur le plan clinique. La méthode statistique choisie doit concorder à la conception de l’étude et au calcul de la taille de l’échantillon effectué pour garantir que les analyses prévues seront suffisamment puissantes pour détecter une différence significative entre les groupes d’étude pour fournir une réponse fiable aux questions posées (par exemple, « Est-ce que le nouveau produit d’essai est non inférieur au produit témoin? » ou « Est-ce que le nouveau produit d’essai est supérieur au produit témoin? »). La puissance de l’étude et le niveau de signification statistique doivent être déclarés avec le taux d’attrition attendu, qui doit être pris en compte dans le calcul de la taille de l’échantillon. Le calcul de la puissance statistique à la fin de l’étude est également requis (23). Le calendrier de toutes les analyses intermédiaires prévues, le cas échéant, ainsi que les critères d’arrêt de l’essai doivent être indiqués.
Les critères d’inclusion et d’exclusion des sujets dans l’analyse par protocole doivent être prévus a priori dans le protocole. Des descriptions détaillées des raisons, de la méthode utilisée et des justifications pour toute exclusion ou rajustement potentiel de données (supprimer une donnée partielle, remplacer des valeurs manquantes, rajuster les valeurs non valides, etc.) de l’analyse doivent être clairement expliquées et décrites dans le rapport d’étude.
Les résultats doivent être indiqués pour les comparaisons entre les groupes d’essai et témoin pour toutes les variables de résultats évaluées. Il faut également comparer les profils de croissance des groupes d’étude avec les références de croissance acceptables pour les prématurés (21). Notamment, les renseignements suivants doivent être fournis (20):
- Pour un essai de non-infériorité, des statistiques descriptives et inférentielles (comme les moyennes, les écarts-types, les intervalles de confiance à 95 % et les mesures de signification statistique) de chaque résultat mesuré pour chaque groupe au début de l’étude et à toutes les étapes d’évaluation tout au long de l’étude doivent être déclarées et analysées. Des analyses par protocole et par intention de traiter doivent être effectuées (20). La non-infériorité et la robustesse des résultats sont clairement établies lorsque l’analyse par protocole et l’analyse par intention de traiter démontrent une absence de différence statistiquement significative entre le groupe recevant la préparation pour prématurés ou le lait humain enrichi d’essai et celui recevant le produit témoin. Dans le cas d’un essai de supériorité, les statistiques ci-dessus pour le résultat principal et les résultats secondaires doivent reposer sur l’analyse par intention de traiter. L’analyse par protocole doit également être effectuée pour évaluer la cohérence des résultats découlant de ces analyses.
- Les estimations de l’ampleur de l’effet (par exemple, la différence de gain de poids entre le groupe d’essai et le groupe témoin) doivent être accompagnées de ses inférences (précisions).
- Nombre de nourrissons qui ont fait l’objet d’analyses à chaque étape, pour chaque analyse.
- Modèles statistiques et covariables utilisés dans l’analyse, avec les justifications appropriées pour leur utilisation.
- Présentation et interprétation des résultats selon l’intervalle de confiance en lien avec la MNI.
- Résultats de l’analyse ajustée et non ajustée.
- Pour chaque sexe, ainsi que pour chaque catégorie de poids à la naissance et d’âge gestationnel, la moyenne du poids corporel, de la taille et de la circonférence de la tête atteints représentée sur les courbes pertinentes, ainsi que leurs intervalles de confiance à 95 % par rapport à l’âge post-menstruel par groupe d’âge.
- Pour chaque sexe, ou catégorie de poids à la naissance ou d’âge gestationnel, le poids, la taille et la circonférence de la tête en fonction de l’âge représentés sur une courbe, les scores z du poids en fonction de la taille et les changements aux scores z, selon les courbes de croissance pour prématurés de Fenton (21), pour chaque groupe d’étude au cours de la période d’étude.
- Nombre total et raisons des abandons ou retraits de nourrissons de l’étude par des chercheurs pour chaque groupe d’étude, ainsi qu’une évaluation ou une explication des répercussions des abandons ou des retraits sur les résultats de l’étude.
- S’il est prévu d’effectuer une analyse de sous-groupe ou d’apporter des rajustements pour tenir compte des facteurs de confusion à l’étape d’analyse, les détails doivent être précisés a priori dans le protocole.
Traitement des données manquantes
Si l’imputation (remplacement des valeurs manquantes par des estimations) des données manquantes est prévue, il faut déclarer l’information sur la robustesse des hypothèses. Le rapport d’étude doit inclure des explications détaillées sur la façon dont ces estimations ou dérivations ont été calculées, le nombre et les raisons des valeurs manquantes dans chaque groupe, les hypothèses sous-jacentes qui ont été émises, et les répercussions des imputations sur les conclusions de l’étude.
Comparaisons multiples
Pour les essais où un rajustement de plusieurs comparaisons est nécessaire, l’approche prévue au préalable pour l’ajustement de la multiplicité doit être précisée.
3.6 Résultats de l’étude
Les résultats de l’étude doivent être déclarés de manière transparente et reproductible, conformément à l’énoncé CONSORT (14).
3.6.1 Organigramme des participants et analyse des retraits et des abandons
Un organigramme est fortement recommandé, car il est utile pour illustrer le cheminement des participants tout au long de l’étude, du recrutement à la fin. Toute déviation du protocole prévu doit être décrite, et les raisons indiquées. Pour chaque groupe de l’étude, il faut indiquer les renseignements détaillés suivants dans l’organigramme :
- nombre total de sujets randomisés, qui reçoivent l’intervention prévue, qui terminent le protocole d’étude, et qui font l’objet d’analyses pour le résultat principal et les résultats secondaires;
- nombre total de sujets qui se retirent ou qui abandonnent l’étude et qui sont exclus des analyses du résultat principal et des résultats secondaires.
Un tableau présentant les caractéristiques démographiques et cliniques de base de chaque groupe doit être inclus.
3.6.2 Analyses de départ
Inclure les résultats des analyses comparant les caractéristiques sociodémographiques et les paramètres anthropométriques à la naissance et au début de l’essai entre les groupes d’essai et témoin dans le rapport.
3.6.3 Analyses du résultat principal et des résultats secondaires
Essai de non-infériorité : inclure les résultats découlant de l’analyse par protocole et de l’analyse par intention de traiter des paramètres du résultat principal et des résultats secondaires (figurant dans la section « Mesure des résultats ») entre le groupe d’essai et le groupe témoin dans le rapport final.
Essai de supériorité : Inclure les résultats découlant de l’analyse par intention de traiter des paramètres du résultat principal et des résultats secondaires entre le groupe d’essai et le groupe témoin dans le rapport final.
Notamment, il faut relever clairement les résultats suivants :
- nombre de participants analysés à chaque moment prédéfini dans l’analyse selon le protocole et l’analyse par intention de traiter;
- statistiques descriptives, y compris la moyenne, la médiane et ses inférences pour chaque variable des résultats selon le sexe et les catégories de poids à la naissance et d’âge gestationnel et selon le groupe d’étude;
- scores z moyens et changements aux scores z pour le poids, la taille, et la circonférence de la tête en fonction de l’âge, le poids selon la taille, et ses inférences par sexe et par catégorie de poids à la naissance et d’âge gestationnel et groupe d’étude au moyen des courbes de croissance pour prématurés de Fenton (21);
- poids, taille et circonférence de la tête atteints à chaque visite de l’étude et au cours de sa durée par sexe et par catégorie de poids à la naissance et d’âge gestationnel, et par groupe d’étude;
- estimations des différences moyennes, de leurs inférences et des valeurs p pour chaque résultat évalué à l’échelle des groupes ci-dessus, ainsi que des discussions détaillées concernant les différences significatives.
Les courbes de croissance pour prématurés de Fenton (21) devraient être utilisées pour comparer les moyennes du groupe d’étude pour le poids, la taille et la circonférence de la tête atteints, ainsi que les scores Z calculés à chaque visite effectuée pendant l’étude, et sur toute la durée de celle-ci.
- Courbes de croissance individuelles
Les données anthropométriques individuelles doivent être représentées sur les courbes de croissance pour prématurés de Fenton (21) afin de tracer la performance de croissance de chaque individu. Les courbes de croissance individuelles doivent être fournies dans la soumission préalable à la mise en marché de la préparation pour prématurés ou du fortifiant pour lait humain.
- Scores Z
Les mesures anthropométriques individuelles devraient être transformées en scores z selon les courbes de croissance pour prématurés de Fenton (21). Ces scores z indiquent la distance et l’orientation d’une observation par rapport à la moyenne de la population (écarts-types supérieurs et inférieurs à la moyenne).
Scores z des groupes : Les scores z pour le poids, la taille et la circonférence de la tête en fonction de l’âge et du poids selon la taille pour chaque groupe caractérisé par sexe, et par catégorie de poids à naissance et d’âge gestationnel doivent être comparés et soumis dans les notifications préalables à la mise en marché.
On peut télécharger des calculatrices des scores z de Fenton à la page www.ucalgary.ca/fenton.
Comme mentionné ci-dessus, dans le cas d’une étude de suivi à long terme, les Normes de croissance de l’enfant de l’Organisation mondiale de la Santé (3) devraient être utilisées pour évaluer la croissance sur une durée d’étude supérieure à 50 semaines d’âge post-menstruel.
3.6.4 Analyses des événements indésirables
Des renseignements détaillés, figurant dans la section des événements indésirables (3.5.11), pour les sujets ayant des incidents d’événements indésirables devraient être interprétés en comparant la manifestation d’événements indésirables dans chaque groupe d’étude et les retraits découlant de ces événements, et aussi en évaluant la gravité par rapport au nombre d’événements. Il est fortement recommandé que l’évaluation clinique des événements indésirables et son imputation au traitement attribué se fassent à l’insu, et dans la mesure du possible, par des professionnels de la santé indépendants. Ces données devraient permettre une évaluation équilibrée des risques et des bénéfices.
Si des examens supplémentaires sont nécessaires pour évaluer la relation entre un événement indésirable grave et l’intervention, tous les examens pertinents ou conclusions de laboratoire doivent être indiqués avec leurs résultats dans un FEC ou dans une pièce jointe à un dossier de suivi.
3.7 Discussion et conclusions
La discussion et l’interprétation des résultats de l’étude doivent bien faire la distinction entre les résultats de signification clinique et statistique. Les conclusions des auteurs doivent être conformes aux résultats déclarés de l’étude en tenant compte des hypothèses de non-infériorité ou de supériorité, et définir clairement le résultat principal et y accorder la priorité. Il convient d’indiquer si les conclusions se rapportant à la non-infériorité ou à la supériorité sont fondées sur une analyse par protocole ou une analyse par intention de traiter, ou les deux, et si ces conclusions sont cohérentes avec les deux analyses pour les essais de non-infériorité. Les conclusions doivent inclure une évaluation équilibrée des bénéfices et des risques, en mettant l’accent sur les limites de l’étude, et doivent tenir compte de toute autre preuve pertinente, y compris la cohérence ou l’incompatibilité par rapport à d’autres essais, les sources de biais potentiel ou le manque de précisions, les différentes normes de soins et les protocoles cliniques standard dans les USIN, insu partiel ou absence d’insu, dangers associés à la multiplicité des analyses et des résultats, et caractère général (validité externe, applicabilité des résultats de l’étude).
4.0 Normes éthiques pour les chercheurs
4.1 Consentement éclairé (Conférence internationale d’harmonisation [ICH], E 6 [R1])
Chaque sujet potentiel (parents ou soignants) doit être bien informé des buts, des méthodes, des sources de financement, de tout conflit d’intérêts possible, des affiliations institutionnelles des chercheurs, des bénéfices prévus et des risques potentiels de l’étude, ainsi que de l’inconfort qui pourrait en découler. Il doit également être mis au courant des dispositions relatives au suivi après l’étude et de tous les autres aspects pertinents de l’étude. Le sujet potentiel doit être informé de son droit de refuser de participer à l’étude ou de retirer son consentement à la participation à tout moment sans représailles (43).
L’étude doit être menée conformément aux principes et aux règles éthiques qui trouvent leur origine dans la déclaration de Helsinki et de ses modifications subséquentes, et doit respecter les BPC ainsi que toutes les exigences réglementaires pertinentes.
Le protocole, toute modification ainsi que le processus de consentement éclairé doivent recevoir l’approbation ou l’avis favorable du comité d’évaluation institutionnel avant le début de l’essai. L’institution qui accorde l’approbation doit être indiquée. Le nom de tout organisme de recherche sous contrat auquel on a demandé d’effectuer le travail doit être fourni.
Les produits expérimentaux doivent être fabriqués, manipulés et stockés conformément aux bonnes pratiques de fabrication applicables. Ils doivent être utilisés conformément au protocole approuvé.
4.2 Financement, parrainage et conflit d’intérêts
Le protocole doit inclure des renseignements concernant le financement, les promoteurs, les affiliations institutionnelles, les conflits d’intérêts possibles, les incitatifs pour les sujets, et l’information concernant les dispositions pour le traitement ou la rémunération des sujets qui subissent des préjudices à la suite de leur participation à l’étude de recherche. Il faut divulguer le rôle exact et les contributions du bailleur de fonds et du promoteur (par exemple, dans la conception, la réalisation, l’analyse et la production de rapports) (18).
Un chercheur, qui est idéalement indépendant de l’industrie des préparations pour nourrissons, doit assumer la responsabilité globale de la réalisation de l’essai, de la planification et de l’exécution des analyses statistiques, ainsi que de la décision de publier, et de la déclaration et de l’interprétation des conclusions de l’essai (18).
4.3 Compétences des chercheurs
Le protocole doit décrire les qualifications et l’expérience que les chercheurs doivent avoir.
4.4 Études de suivi à long terme
Il est encouragé de recueillir les données cliniques par des études de suivi à long terme (c.-à-d. jusqu’à 20 mois d’âge corrigé) chez les prématurés, car cela permettra de mieux comprendre la qualité de la croissance et de se pencher sur la façon d’équilibrer la prise de poids pour optimiser les résultats du développement neurologique par rapport au risque de maladies cardiovasculaires et métaboliques précoces (36, 46).
On pourrait évaluer les paramètres à long terme suivants :
- résultats liés au développement, comme les résultats sur l’échelle Bayley ou aux tests de quotient intellectuel, habituellement évalués après 20 mois d’âge corrigé;
- évaluation de la composition corporelle, par exemple, changements à la masse maigre et à la masse grasse.
Remarque : Compte tenu des progrès et de l’évolution du domaine de la gestion nutritionnelle des prématurés et des nourrissons de faible poids à la naissance, ce protocole sera mis à jour à l’avenir en fonction des nouvelles données de recherche.
5.0 Glossaire et références
5.1 Glossaire
Alimentation trophique ou nutrition entérale minimale
Petit volume de lait donné par alimentation entérale au cours des 24 premières heures.
Analyse ajustée
Terme qui renvoie habituellement aux tentatives de tenir compte des inégalités des données de référence entre les groupes pour les caractéristiques importantes des patients. Renvoie parfois au rajustement de la valeur p pour prendre en compte des tests multiples (42)
Analyse par intention de traiter
Stratégie pour analyser les données d’un essai contrôlé randomisé. Tous les participants sont inclus dans le groupe auquel ils ont été affectés, peu importe s’ils ont reçu (ou terminé) ou non l’intervention donnée à ce groupe. L’analyse par intention de traiter prévient le biais causé par la perte de participants, qui peut perturber l’équivalence de départ établie par la randomisation et qui peut refléter le non-respect du protocole. Ce terme est souvent utilisé à tort dans les publications d’essais lorsque certains participants ont été exclus (48).
Analyse par protocole
Analyse du sous-ensemble de participants d’un essai contrôlé randomisé qui se sont suffisamment conformés au protocole pour garantir que leurs données seraient susceptibles de démontrer l’effet du traitement. Ce sous-ensemble peut être défini après avoir pris en compte l’exposition au traitement, la disponibilité des mesures et l’absence de violations majeures du protocole. La stratégie d’analyse par protocole peut être sujette à un biais car les raisons de la non-conformité peuvent être liées au traitement (48).
Attrition
Perte de participants au cours de l’essai (également appelé « perte lors du suivi »). Les participants qui sont perdus au cours de l’essai sont souvent appelés des « abandons » (48).
Biais
Distorsion systématique de l’effet d’intervention estimé par rapport à la « vérité » causée par des lacunes dans la conception, la réalisation ou l’analyse d’un essai (42).
Biais de sélection
Erreur systématique dans la création de groupes d’interventions, qui provoque des différences de pronostic entre eux, c’est-à-dire que les caractéristiques de base mesurées ou non des groupes diffèrent en raison de la façon dont les participants ont été sélectionnés pour l’étude ou affectés à leur groupe d’étude. Ce terme est également utilisé pour indiquer que les participants ne sont pas tous représentatifs de la population de tous les participants possibles (45).
Caractère généralisable, généralisation
Mesure dans laquelle les résultats d’un essai clinique peuvent être extrapolés de manière fiable, à partir des sujets qui ont participé à l’essai à une population plus vaste de patients et à un éventail plus large de contextes cliniques (45).
Classement des événements indésirables comme étant légers, modérés et sévères
Outil souvent utilisé dans les études non oncologiques. La définition de léger, modéré et sévère peut être différente d’un protocole d’étude à un autre. La sévérité (intensité) de chaque événement indésirable, y compris les événements indésirables graves, est attribuée à l’une des catégories suivantes :
- Léger – Événement facilement tolérable par le sujet, qui cause peu d’inconfort et qui ne nuit pas aux activités quotidiennes
- Modéré – Événement suffisamment gênant pour nuire aux activités quotidiennes normales
- Sévère – Événement qui empêche les activités quotidiennes normales
OU
- Léger – Conscience du signe ou de symptôme, mais celui-ci est facilement toléré
- Modéré – Inconfort suffisant pour nuire aux activités normales
- Sévère – Symptôme invalidant qui rend impossible la réalisation des activités normales (42)
Déviation du protocole
Une déviation du protocole se produit lorsque, sans conséquences importantes, les activités d'une étude s'écartent du protocole approuvé par le comité d'examen institutionnel, par exemple, en manquant une fenêtre de visite parce que le sujet est en voyage. Cette situation n'est pas aussi grave qu'une violation de protocole (44).
Dissimulation de la répartition
Technique utilisée pour garantir une séquence imprévisible des attributions. Il s’agit d’un processus critique qui empêche de connaître à l’avance le traitement attribué, et permet donc à ceux qui inscrivent les participants de ne pas être influencés par cette information. La décision d’accepter ou de rejeter un participant doit être prise, et un consentement éclairé doit être obtenu du participant, dans l’ignorance de la prochaine attribution dans la séquence (47).
Écart-type
Mesure de l’étendue ou de la dispersion d’un ensemble d’observation, calculée comme la différence moyenne par rapport à la valeur moyenne de l’échantillon (48).
Effet indésirable
Événement indésirable pour lequel il existe tout au moins une possibilité raisonnable qu’il y ait un lien de causalité entre l’intervention et l’événement (42).
Efficacité potentielle
Mesure dans laquelle une intervention donne un résultat bénéfique dans des conditions idéales. Les essais cliniques qui évaluent l’efficacité potentielle sont parfois appelés des essais explicatifs et sont limités aux participants qui coopèrent pleinement (48).
Efficacité potentielle vs efficacité réelle
Les études d’intervention peuvent être placées sur une échelle, avec une progression allant d’essais d’efficacité potentielle à des essais d’efficacité réelle. L’efficacité potentielle peut être définie comme la performance d’une intervention dans des circonstances idéales et contrôlées, alors que l’efficacité réelle renvoie à sa performance dans des conditions réelles (50).
Enrichissement standard
Méthode d’enrichissement la plus utilisée. La pratique courante consiste à ajouter une quantité fixe de fortifiant composé par 100 mL de lait humain pour atteindre les apports nutritionnels recommandés. Cette quantité fixe a été calculée et déterminée par le fabricant en supposant une teneur en protéines fixe pour tous les échantillons de lait, sans tenir compte des variations intra-sujets et inter-sujets et des variations temporelles. L’enrichissement standard commence habituellement lorsque le volume de lait donné est de 50 à 100 ml/kg. Selon le consensus de la réunion conjointe de EMBA/ESPGHAN/AAP de Milan, il est recommandé d’enrichir le lait humain pour les prématurés ayant un poids à la naissance inférieur à 1 800 g (53).
Essai clinique
Expérience visant à comparer les effets de deux ou plusieurs interventions en matière de soins de santé. L’expression « essai clinique » est un terme générique qui désigne des essais sur les soins de santé menés selon différentes méthodologies, notamment les essais non contrôlés, les essais contrôlés et les essais contrôlés randomisés (48).
Essai de non-infériorité
Essai dont l’objectif principal est de montrer que la réponse à un produit expérimental n’est pas cliniquement inférieure à un produit de référence (témoin actif ou placebo), une version unilatérale d’un essai d’équivalence (48).
Essai de supériorité
Essai dont l’objectif principal est de montrer que la réponse au produit expérimental est supérieure à un produit de référence (témoin actif ou placebo) (45).
Essai multicentrique
Essai clinique mené selon un seul protocole mais à différents emplacements, et donc, mené par plus d’un chercheur (45).
Erreur de type I
Conclusion selon laquelle un traitement fonctionne, alors que ce n’est pas réellement le cas. Le risque d’une erreur de type 1 est souvent appelé alpha. Dans un test statistique, il décrit la probabilité de rejeter l’hypothèse nulle alors qu’elle est en fait vraie (aussi appelé un « faux positif ») (45).
Erreur de type II
Conclusion selon laquelle aucune preuve n’indique qu’un traitement fonctionne, alors qu’en réalité, le traitement fonctionne. Le risque d’une erreur de type 2 est souvent appelé bêta. Dans un test statistique, il décrit la probabilité de ne pas rejeter l’hypothèse nulle alors qu’elle est en fait fausse. Le risque d’une erreur de type II diminue à mesure que le nombre de participants à une étude augmente (on parle aussi de faux négatif) (48).
Événement indésirable
Résultat indésirable qui survient pendant ou après l’administration d’une drogue ou une autre intervention, mais qui n’est pas nécessairement causé par la drogue ou l’intervention (42).
Événement indésirable grave (dans le contexte d’un essai clinique)
Événement qui, de l’avis du chercheur ou du promoteur, cause l’un des résultats suivants : décès, événement indésirable mettant la vie en danger, hospitalisation nouvelle ou prolongée, incapacité persistante ou importante, ou perturbation importante de la capacité de mener les fonctions normales de la vie (43).
Facteur de confusion
Facteur qui est associé à une intervention (ou à une exposition) ainsi qu’au résultat d’intérêt (48).
Hypothèse nulle
En termes très simples, l’hypothèse nulle suppose que le facteur d’intérêt (par exemple, traitement) n’a aucune incidence sur le résultat (par exemple, risque de décès) (48).
Imputation
Procédure consistant à saisir une valeur pour un élément de données spécifique lorsque la réponse est manquante ou inutilisable. Cette procédure consiste à modifier certaines réponses ou à attribuer des valeurs lorsqu'elles sont manquantes dans l'enregistrement en cours d'édition afin de garantir la qualité des estimations et la création d'un enregistrement plausible et cohérent en interne (50).
Indice d’Apgar
Indice objectif de l’état du bébé après la naissance. Il est déterminé en évaluant la fréquence cardiaque, l’effort respiratoire, le tonus musculaire, la couleur de la peau et la réaction à un cathéter dans la narine. Pour chacun de ces signes objectifs, on attribue zéro, un ou deux points. Un indice d’Apgar de 10 signifie que le nourrisson est dans le meilleur état possible. Cette évaluation se fait de façon systématique 60 secondes après la naissance du nourrisson. Un enfant ayant un indice entre zéro et trois doit être réanimé immédiatement. On refait souvent cette évaluation cinq minutes après la naissance, et en cas de réanimation difficile, on peut déterminer de nouveau l’indice d’Apgar après 10, 15 et 20 minutes.
Innocuité
Preuves substantielles de l’absence de risques. Ce terme est souvent utilisé à tort lorsqu’il n’y a tout simplement pas de preuve de risques (42).
Insu
Processus consistant à empêcher ceux qui participent à un essai de savoir à quel groupe de comparaison appartient un participant particulier. Pratique qui consiste à faire en sorte que les participants à l’essai, les prestataires de soins, les collecteurs de données, et parfois ceux qui analysent les données, ne sachent pas quelle intervention est administrée à quel participant (42).
Intervalle de confiance
Mesure de précision d’une valeur estimée. L’intervalle représente la plage de valeurs, en toute cohérence avec les données qui sont présumées englober la valeur « véritable » avec une probabilité élevée (habituellement 95 %). L’intervalle de confiance est exprimé dans les mêmes unités que l’estimation. De grands intervalles indiquent une précision réduite, et des intervalles étroits, une plus grande précision (49).
Marge de non-infériorité
La marge est la plus grande différence qui peut être jugée cliniquement acceptable; elle devrait être inférieure aux différences observées dans les essais de supériorité du comparateur actif (45).
Moyenne
Valeur moyenne, calculée en ajoutant toutes les observations et en divisant par le nombre d’observations (48).
Multiplicité
Prolifération des comparaisons possibles dans un essai. Les sources courantes de multiplicité sont les résultats multiples, les résultats évalués à plusieurs moments après l’intervention, les analyses de sous-groupes ou les groupes d’intervention multiples (49).
Précision [en statistiques]
Probabilité d’erreurs aléatoires dans les résultats d’une étude, d’une méta-analyse ou d’une mesure. Plus la précision est grande, moins il y a d’erreurs aléatoires. Les intervalles de confiance autour de l’estimation de l’effet de chaque étude sont une façon d’exprimer la précision; un intervalle de confiance plus étroit signifie plus de précision (48).
Puissance
Probabilité qu’un essai détectera comme étant statistiquement significatif un effet d’intervention d’une taille précisée. La taille avant l’essai est souvent choisie pour donner à l’essai la puissance voulue (45).
Réaction indésirable
Événements pour lesquels un lien de causalité avec l’intervention est bien établi et suffisamment solide (sensible et précis) pour justifier l’attribution de l’événement à l’intervention (42).
Résultat principal
Résultat jugé comme étant le plus important (48).
Score Z
Écart de la valeur pour un individu par rapport à la valeur moyenne de la population de référence divisée par l’écart-type pour la population de référence (52).
Séquence de répartition
Liste d’interventions, classées de façon aléatoire, utilisées pour affecter des participants inscrits en ordre séquentiel à des groupes d’intervention (42).
Témoin
- [Dans un essai contrôlé :] Un participant dans le groupe qui sert de comparateur pour une ou plusieurs interventions expérimentales. Les sujets du groupe témoin peuvent recevoir un placebo, aucun traitement, un traitement standard ou une intervention active, comme un médicament standard.
- [Dans les statistiques :] Rajuster en fonction des observations ou des influences extérieures ou en tenir compte (48).
Test d’hypothèse
Procédure statistique pour déterminer s’il faut rejeter une hypothèse nulle sur la base des données observées (48).
Valeur p
La probabilité (allant de presque zéro à un) que les résultats observés dans une étude (ou des résultats plus extrêmes) aient pu se produire par hasard si, en réalité, l’hypothèse nulle était vraie.
Validité externe
Mesure dans laquelle les résultats d’un essai constituent une base correcte pour la généralisation à d’autres circonstances (42).
Violation du protocole
Une divergence par rapport au protocole qui : réduit matériellement la qualité ou l'exhaustivité des données, rend le formulaire de consentement éclairé inexact ou a un impact sur la sécurité, les droits ou le bien-être d'un sujet (44).
5.2 Références
- American Academy of Pediatrics, Committee on Fetus and Newborn. (2004). Age terminology during the perinatal period. Pediatrics, 114 (5), 1362-1364.
- World Health Organization (WHO). (2018). Naissances prématurées: Principal repère. [En ligne]. Accès: https://www.who.int/fr/news-room/fact-sheets/detail/preterm-birth
- World Health Organization (WHO). (2006). Optimal feeding of low-birth-weight infants. [En ligne, anglais seulement]. Accès: https://apps.who.int/iris/bitstream/handle/10665/43602/9789241595094_eng.pdf;jsessionid=80736F92F581729831081B1CA71D704B?sequence=1
- Groh-Wargo S, Thompson M, Cox H (eds). (2000). Nutritional Care for High-Risk Newborns. Precept Press, Inc., Chicago, IL.
- Goldenberg R, Culhane JF, Iams JD, Romero R. (2008). Epidemiology and causes of preterm birth. The Lancet, 371(9606), 75-84.
- Malhotra N, Puri R, Malhotra J, Chervenak FA, Kurjak A (eds). (2012). Donald School Manual of Practical Problems in Obstetrics, Jaypee Brothers Medical Publishers.
- Sharma D, Shastri S, Sharma P. (2016). Intrauterine Growth Restriction: Antenatal and Postnatal Aspects. Clinical Medicine Insights: Pediatrics. 10, 67-83.
- Goldberg D, Becker PJ, Brigham K, Carlson S, Fleck L, Gollins L, Sandrock M, Fullmer M, Van Poots HA. (2018). Identifying Malnutrition in Preterm and Neonatal Populations: Recommended Indicators. J Acad Nutr Diet. 118(9),1574-1582.
- Cormack BE, Embleton ND, van Goudoever JB, Hay WW Jr, Bloomfield FH. (2016). Comparing apples with apples: it is time for standardized reporting of neonatal nutrition and growth studies. Pediatr Res. 79(6):810-20.
- Patel AL, Engstrom JL, Meier PP, Kimura RE. (2005). Accuracy of methods for calculating postnatal growth velocity for extremely low birth weight infants. Pediatrics. 116(6):1466-73.
- Patel AL, Engstrom JL, Meier PP, Jegier BJ, Kimura RE. (2009). Calculating postnatal growth velocity in very low birth weight (VLBW) premature infants. J Perinatol. 29(9):618-22.
- Fenton TR, Senterre T, Griffin IJ. (2019). Time interval for preterm infant weight gain velocity calculation precision. Arch Dis Child Fetal Neonatal Ed. 104(2):F218-F219.
- Fenton TR, Griffin IJ, Hoyos A, Groh-Wargo S, Anderson D, Ehrenkranz A, Senterre T. (2019). Accuracy of preterm infant weight gain velocity calculations vary depending on method used and infant age at time of measurement. Pediatr Res., 85, 650-654.
- Schulz KF, Altman DG, Moher D; CONSORT Group. (2010). CONSORT 2010 Statement: Updated guidelines for reporting parallel group randomised trials. J Clin Epidemiol. 63(8), 834-40.
- World Health Organization (WHO). (2021). WHO primary registries. [En ligne, anglais seulement]. Accès : https://www.who.int/clinical-trials-registry-platform/network/primary-registries.
- World Health Organization. (2009). WHO Registry Criteria (Version 2.1, April 2009). [En ligne, anglais seulement]. Accès : https://www.who.int/clinical-trials-registry-platform/network/registry-criteria.
- Boutron I, Altman DG, Moher D, Schulz KF, Ravaud P; CONSORT NPT Group. (2017). CONSORT Statement for Randomized Trials of Nonpharmacologic Treatments: A 2017 Update and a CONSORT Extension for Nonpharmacologic Trial Abstracts. Ann Intern Med. 67(1):40-47.
- Jarrold K, Helfer B, Eskander M, Crawley H, Trabulsi J, Caulfield LE, Duffy G, Garcia-Larsen V, Hayward D, Hyde M, Jeffries S, Knip M, Leonardi-Bee J, Loder E, Lodge CJ, Lowe AJ, McGuire W, Osborn D and Przyrembel H. (2020). Guidance for the conduct and reporting of clinical trials of breast milk substitutes. JAMA Pediatr, 174(9):874-881.
- Piaggio G, Elbourne DR, Pocock SJ, Evans SJ, Altman DG; CONSORT Group. (2012). Reporting of noninferiority and equivalence randomized trials: extension of the CONSORT 2010 statement. JAMA. 308(24), 2594-604.
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies). (2017). Scientific and technical guidance for the preperation and presentation of an application for authorisation of an infant and/or follow-on formula manufacturer from protein hydrolysates. EFSA Journal, 15(5), 4779.
- Fenton TR and Kim J. (2013). A systematic review and meta-analysis to revise the Fenton growth chart for preterm infants. BMC Pediatrics, 13(59).
- Fenton TR, Anderson D, Groh-Wargo S, Hoyos A, Ehrenkranz RA, Senterre T. (2018). An attempt to standardize the calculation of growth velocity of preterm infants—evaluation of practical bedside methods. The Journal of Pediatrics, 196,77-83.
- Food and Drug Adminstration. (2014). Code of Federal Regulations Title 21, Chapter I, Subchapter B, Part 106, 106.96 and 106.121. [En ligne, anglais seulement]. Accès : https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=106.
- Kleinman R. (2019). Pediatric Nutrition, 8th Edition, American Academy of Pediatrics.
- Viswanathan S, McNelis K, Super D, Einstadter D, Groh-Wargo S, Collin M. (2015). Standardized Slow Enteral Feeding Protocol and the Incidence of Necrotizing Enterocolitis in Extremely Low Birth Weight Infants. JPEN J Parenter Enteral Nutr, 39(6):644-54.
- U.S. Food and Drug Administration. (2016). Guidance Document: Non-Inferiority Clinical Trials," 2016. [En ligne, anglais seulement]. Accès : https://www.fda.gov/media/78504/download
- Rigo J, Hascoët JM, Billeaud C, Picaud JC, Mosca F, Rubio A, Saliba E, Radkë M, Simeoni U, Guillois B, de Halleux V, Jaeger J, Ameye L, Hays NP, Spalinger J. (2017). Growth and Nutritional Biomarkers of Preterm Infants Fed a New Powdered Human Milk Fortifier: A Randomized Trial. J Pediatr Gastroenterol Nutr. 65(4):e83-e93.
- Hair A, Blanco CL, Moreira AG, Hawthorne KM, Lee M, Rechtman DJ, Abrams S. (2014). Randomized trial of human milk cream as a supplement to standard fortification of an exclusive human milk-based diet in infants 750-1250 g birth weight. J Pediatr., 1655(5), 915-20.
- Kim JH, Chan G, Schanler R, Groh-Wargo S, Bloom B, Dimmit R, Williams L, Baggs G, Barrett-Reis B. (2015). Growth and Tolerance of Preterm Infants Fed a New Extensively Hydrolyzed Liquid Human Milk Fortifier. J Pediatr Gastroenterol Nutr., 61(6), 65-71.
- Lucas A, Fewtrell MS, Morley R, Lucas PJ, Baker BA, Lister G, Bishop NJ. (1996). Randomized outcome trial of human milk fortification and developmental outcome in preterm infants. Am J Clin Nutr., 64(2), 142-51.
- Moya F, Sisk P, Walsh KR, Berseth CL. (2012). A new liquid human milk fortifier and linear growth in preterm infants. Pediatrics, 130(4), e928-35.
- Schanler R, Groh-Wargo SL, Barrett-Reis B, White RD, Ahmad K, Oliver J, Baggs G, Williams L, Adamkin D. (2018). Improved Outcomes in Preterm Infants Fed a Nonacidified Liquid Human Milk Fortifier: A Prospective Randomized Clinical Trial. J Pediatr., 202, 31-37.
- Chen L and Nguyen L. Meta-Analysis for non-inferiority margins of weigh gains between formula-fed and breast-fed infant. manuscript in preparation.
- Quigley M and McGuire W. (2018). Formula versus donor breast milk for feeding preterm or low birth weight infants. Cochrane Database Syst Rev., 6(6).
- Brown J, Lin L, Embleton ND, Harding JE, McGuire W. (2020). Multi-nutrient fortification of human milk for preterm infants., Cochrane Database of Systematic Reviews, 6.
- Tyson J and Ahn C. (2002). Evaluation of the growth of preterm or low birth weight infants. Food Advisory Committee on Infant Formula, Food and Drug Administration.
- Schumi J and Wittes J. (2011). Through the looking glass: understanding non-inferiority. Trials, 12(106), 1-12.
- The European Agency for the Evaluation of Medicinal Products Evaluation of Medicines for Human Use. (2000). Points to consider on switching between superiority and non-inferiority. [En ligne, anglais seulement]. Accès : https://www.ema.europa.eu/en/documents/scientific-guideline/points-consider-switching-between-superiority-non-inferiority_en.pdf.
- Moher D, Hopewell S, Schulz KF, Montori V, Gøtzsche PC, Devereaux PJ, Elbourne D, Egger M, Altman DG. (2010). CONSORT 2010 Explanation and Elaboration: updated guidelines for reporting parallel group randomised trials. Journal of Clinical Epidemiology, 8,63.
- Arslanoglu, S., Boquien, C. Y., King, C., Lamireau, D., Tonetto, P., Barnett, D., Bertino, E., Gaya, A., Gebauer, C., Grovslien, A., Moro, G. E., Weaver, G., Wesolowska, A. M., & Picaud, J. C. (2019). Fortification of Human Milk for Preterm Infants: Update and Recommendations of the European Milk Bank Association (EMBA) Working Group on Human Milk Fortification. Frontiers in Pediatrics, 7(76).
- Institute of Medicine (US) Committee on the Evaluation of the Addition of Ingredients New to Infant Formula. (2004). Infant formula: evaluating the safety of new ingredients. Washington (DC). National Academies Press.
- Ioannidis JPA, Evans SJW, Gøtzsche PC, O'Neill RT, Altman DG, Schulz K, Moher D for the CONSORT group. (2004). Better reporting of harms in randomized trials: an extension of the CONSORT statement. Ann Intern Med, 141(10), 781-8.
- International Conference on Harmonisation (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use. (2016). ICH harmonised tripartite guideline for good clinical practice E6 (R2). [En ligne, anglais seulement]. Available: https://database.ich.org/sites/default/files/E6_R2_Addendum.pdf
- Bhatt. (2012). Protocol deviation and violation. Perspectives in Clinical Research, 3(3), 117.
- International Conference on Harmonisation (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use. (1998). ICH Harmonised Tripartite Guideline, Statistical Principles For Clincal Trials E9. [En ligne, anglais seulement]. Accès : https://database.ich.org/sites/default/files/E9_Guideline.pdf.
- Raiten DJ, Steiber AL, Carlson SE, Griffin I, Anderson D, Hay WW Jr, Robins S, Neu J, Georgieff MK, Groh-Wargo S, Fenton TR; Pre-B Consultative Working Groups. (2016). Working group reports: evaluation of the evidence to support practice guidelines for nutritional care of preterm infants-the Pre-B Project. Am J Clin Nutr. 103(2):648S-78S
- Deng C. (2018). Grading the severity of AEs and its impact on AE reporting. [En ligne, anglais seulement]. Accès : http://onbiostatistics.blogspot.com/2018/05/grading-severity-of-aes-and-its-impact.html.
- Cochrane Collaboration. (2019). Glossary of Cochrane Terms [En ligne, anglais seulement]. Accès : https://epoc.cochrane.org/sites/epoc.cochrane.org/files/public/uploads/SURE-Guides-v2.1/Collectedfiles/source/glossary.html.
- Altman DG, Schulz KF, Moher D, Egger M, Davidoff F, Elbourne PC, GØtzsche PC, Lang T for the CONSORT Group. (2001). The revised CONSORT statement for reporting randomized trials: explanation and elaboration. Annals of internal medicine, 134(8), 663-694.
- Singal AG, Higgins PDR, Waljee A. (2014). A Primer on Effectiveness and Efficacy Trials, Clin Transl Gastroenterol., 5(1),e45.
- Organisation for Economic Co-operation and Development. (2020). Economic Commission of Europe of the United Nations (UNECE) Glossary of terms on statistical data editing, conference of European statisticians, methodological material. [En ligne, anglais seulement]. Accès : https://stats.oecd.org/glossary/detail.asp?ID=3462.
- Centers for Disease Control, National Center for Health Statistics. [En ligne, anglais seulement]. Accès : https://www.cdc.gov/nchs/index.htm.
- Moro GE, Arslanoglu S, Bertino E, Corvaglia L, Montirosso R, Picaud JC, Polberger S, Schanler RJ, Steel C, van Goudoever J, Ziegler E. (2015). Human milk in feeding premature infants: consensus statement. Journal of Pediatric Gastroenterology and Nutrition, 61(1), S16-S19.
Appendice 1: Liste de contrôle CONSORT 2010 des renseignements à inclure dans les rapports d’un essai randomiséNote de bas de page 1
|
Section ou sujet |
No du point |
Points de la liste de contrôle |
|---|---|---|
| Titre | 1a | Indication d’un essai randomisé dans le titre |
| Résumé | 1b | Résumé structuré du plan de l’essai, des méthodes, des résultats et des conclusions (pour des conseils spécifiques, voir CONSORT pour les résumés (21,31)) |
|
Introduction |
||
|
Contexte et objectifs |
2a | Renseignements scientifiques et explication de la justification |
| 2b | Objectifs ou hypothèses précis | |
|
Méthodes |
||
|
Plan de l’essai |
3a | Description du plan de l’essai (parallèle, factoriel, etc.), y compris le rapport de répartition |
| 3b | Changements importants aux méthodes après le début de l’essai (comme les critères d’admissibilité) avec des raisons à l’appui | |
|
Participants |
4a | Critères d’admissibilité des participants |
| 4b | Paramètres et endroits où les données ont été recueillies | |
| Interventions | 5 | Interventions pour chaque groupe avec suffisamment de détails pour permettre leur reproduction, y compris comment et quand elles ont été effectivement administrées |
|
Résultats |
6a | Mesures du résultat principal et des résultats secondaires préalablement précisées et complètement définies, y compris comment et quand elles ont été évaluées |
| 6b | Tout changement dans les résultats de l’essai après le début de celui-ci, avec les raisons | |
|
Taille de l’échantillon |
7a | Comment la taille de l’échantillon a été déterminée |
| 7b | Le cas échéant, explication des analyses intermédiaires et des directives d’arrêt | |
|
Randomisation |
||
|
Génération de séquence |
8a | Méthode utilisée pour générer la séquence de répartition aléatoire |
| 8b | Type de randomisation; détails de toute restriction (comme le blocage et la taille des blocs) | |
| Mécanisme de dissimulation de la répartition | 9 | Mécanisme utilisé pour mettre en œuvre la séquence de répartition aléatoire (comme des conteneurs numérotés de façon séquentielle), décrivant toute mesure prise pour dissimuler la séquence jusqu’à ce que les interventions soient assignées |
| Mise en œuvre | 10 | Qui a généré la séquence de répartition aléatoire, qui a recruté les participants et qui a assigné les participants aux interventions |
|
Insu |
11a | Si c’est le cas, qui était à l’insu après l’affectation aux interventions (par exemple, les participants, les prestataires de soins, les personnes chargées d’évaluer les résultats) et comment |
| 11b | Description de la similitude des interventions, le cas échéant | |
|
Méthodes statistiques |
12a | Méthodes statistiques utilisées pour comparer les groupes pour le résultat principal et les résultats secondaires |
| 12b | Méthodes pour les analyses supplémentaires, telles que les analyses de sous-groupes et les analyses ajustées | |
|
Résultats |
||
|
Flux des participants (un diagramme est fortement recommandé) |
13a | Pour chaque groupe, le nombre de participants qui ont été assignés de manière aléatoire, ont reçu le traitement prévu et ont fait l’objet d’analyses pour le résultat principal |
| 13b | Pour chaque groupe, les pertes et les exclusions après la randomisation, ainsi que les raisons | |
|
Recrutement |
14a | Dates définissant les périodes de recrutement et de suivi |
| 14b | Raison pour laquelle l’essai s’est terminé ou a été interrompu | |
| Données de référence | 15 | Tableau présentant les caractéristiques démographiques et cliniques de base de chaque groupe |
| Chiffres analysés | 16 | Pour chaque résultat principal et secondaire, les résultats pour chaque groupe, ainsi que l’ampleur de l’effet estimé et sa précision (comme l’intervalle de confiance à 95 %) |
|
Résultats et estimation |
17a | Pour chaque résultat principal et secondaire, les résultats pour chaque groupe, ainsi que l’ampleur de l’effet estimé et sa précision (comme l’intervalle de confiance à 95 %) |
| 17b | Pour les résultats binaires, il est recommandé de présenter à la fois les tailles d’effet absolues et relatives | |
| Analyses secondaires | 18 | Résultats de toute autre analyse effectuée, y compris les analyses de sous-groupes et les analyses ajustées, en distinguant les analyses préétablies des analyses exploratoires |
| Risques | 19 | Tous les effets indésirables ou non voulus importants dans chaque groupe (pour des directives précises, consulter CONSORT pour les risques (28)) |
|
Discussion |
||
| Limites | 20 | Limites de l’essai, en abordant les sources de biais potentiel, d’imprécision et, le cas échéant, la multiplicité des analyses |
| Caractère général | 21 | Caractère général (validité externe, applicabilité) des résultats de l’essai |
| Interprétation | 22 | Interprétation cohérente avec les résultats, en mettant en balance les bénéfices et les risques, et en tenant compte d’autres données probantes pertinentes |
|
Autres renseignements |
||
| Enregistrement | 23 | Numéro d’enregistrement et nom du registre d’essai |
| Protocole | 24 | Où le protocole complet de l’essai peut être consulté, s’il est disponible |
| Financement | 25 | Sources de financement et autres mesures de soutien (comme l’approvisionnement en produits pharmaceutiques), rôle des bailleurs de fonds |
Notes de bas de page
- Note de bas de page 1
-
« CONSORT 2010 Statement: updated guidelines for reporting parallel group randomised trials » (14),– Nous recommandons vivement de lire cette déclaration en même temps que le document « CONSORT 2010 Explanation and Elaboration » ou des clarifications importantes sur tous les points.
Appendice 2: Indicateurs biochimiques du bilan protéique et de l’état osseux chez les prématurésNote de bas de page 1
|
Mesure |
Avantages |
Limites |
|---|---|---|
| Bilan protéique: | ||
| Azote uréique sanguin (AUS) | Un faible taux d’AUS est un bon indicateur d’un faible apport en protéines chez les nourrissons cliniquement stables et nourris par voie entérale. | Un taux d’AUS élevé est difficile à interpréter, comme il pourrait indiquer un apport approprié en acides aminés, un faible apport énergétique par rapport à l’apport en protéines ou encore une intolérance aux acides aminés. |
| Préalbumine sérique | Demi-vie d’environ 2 jours. Un faible taux reflète un apport insuffisant en protéines. |
Une inflammation ou une infection peut diminuer le taux de préalbumine. |
| Protéine de liaison du rétinol (PLR) | Demi-vie d’environ 12 h. Un faible taux reflète un apport insuffisant en protéines. |
Les taux de PLR peuvent également être affectés par une teneur sous-optimale de fer, de zinc ou de vitamine A. Mesurer les taux de PLR est plus coûteux que mesurer la préalbumine, tout en fournissant des renseignements équivalents. |
| Transferrine sérique | Marqueur complémentaire du bilan protéique. | En cas de carence en fer, la concentration de transferrine augmente peu importe l’état nutritionnel. Elle est rarement utilisée. |
|
Statut osseux: |
||
| Calcium sérique | Mauvais marqueur d’une maladie métabolique de l’os (MMO). | |
| Phosphate sérique | Haute spécificité et valeur prédictive positive comme marqueur d’une MMO. | Faible sensibilité et valeur prédictive négative comme marqueur d’une MMO. Preuve insuffisante comme marqueur fiable d’une MMO. |
| Phosphatase alcaline sérique | Des niveaux > 900 U/L donnent une spécificité de 71 % et une sensibilité de 88 % comme marqueur d’une MMO. | Preuve insuffisante comme marqueur fiable d’une maladie métabolique de l’os. |
| Phosphatase alcaline sérique et phosphate sérique | Phosphatase alcaline > 900 U/L plus phosphate < 1,8 mmol/L (5,6 mg/dL) donnent une spécificité de 70 % et une sensibilité de 100 % comme marqueur d’une MMO. | Preuve insuffisante comme marqueur fiable d'une MMO. |
| Calcium dans l’urine et marqueurs de phosphate | Le rapport de calcium dans l’urine et de créatinine, la concentration de phosphate et la réabsorption tubulaire de phosphate peuvent être utilisés de façon complémentaire dans le diagnostic d’une MMO. | Calcium dans l’urine et marqueurs de phosphate |
Notes de bas de page
- Note de bas de page 1
-
Pereira-da-Silva, L., Virella, D., & Fusch, C. (2019). Nutritional assessment in preterm infants: a practical approach in the NICU. Nutrients, 11(9), 1999.
Appendice 3. Modèle des besoins nutritifs pour les préparations pour prématurés ou pour les fortifiants avec du lait humain (tels que consommé) par 100 kcal
NOTE: Une valeur unique représente une recommandation minimale. Une valeur entre crochets a été rapportée comme la valeur calculée dans la citation indiquée.
- MACRONUTRIMENTS
Éléments nutritifs |
Exigences du RAD |
Recommandations des experts internationaux |
Profil nutritionnel |
Spécification |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
Nom |
Unité |
ESPGHAN (2010)Référence 1 |
Tsang, et. al. (2005)Référence 2 >1000 g P.C. |
LSRO (2002)Référence 3 |
OMSRéférence 4 >1000 g P.C. (stabilisation à terme)Remarque a |
Koletzko, et. al. (2014)Référence 5 |
Min |
Max |
||
Protéines |
g |
1,8 – 4 |
3,2 – 4,1Remarque b |
2,6 – 3,8 |
2,5 – 3,6 |
[2,5 – 3,0] |
3,2 – 4,1 |
|||
Lipides |
g |
3,3 – 6 |
4,4 – 6,0 |
4,1 – 6,5 |
4,4 – 5,7 |
[3,8 – 5,7] |
4,4 – 6,0 |
|||
- AL |
g |
0,5 |
0,350 – 1,400 |
0,462 – 1,309 |
[0,350 – 1,425]Remarque c |
AP |
0,350 – 1,400 |
|||
- AAL |
g |
AP |
>0,050 |
AP (1 – 4 E% par kg/jour) |
0,077 – 0,228 (1,75 – 4% des acids gras totaux) |
AP |
0.050 |
|||
- AL:AAL |
- |
AP |
5 – 15 |
5 – 15 |
6 – 16 |
AP |
AP |
|||
- AA |
g |
AP |
0,016 – 0,039 |
0,022 |
[0 – 0,034] Remarque d |
AP |
0,032 – 0,041 |
|||
- ADH |
g |
AP |
0,011 – 0,027 |
0,016 |
[0 – 0,020]Remarque e |
AP |
0,050 – 0,055 |
|||
Glucides |
g |
AP |
10,5 – 12 |
5,4 – 15.5 |
9,6 – 12.5 |
[6,3 – 12.9] |
10,5 – 12 |
|||
- VITAMINES ET MINÉRAUX
Éléments nutritifs |
Exigences du RAD |
Recommandations des experts internationaux |
Profil nutritionnel |
Spécification |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
Nom |
Unité |
ESPGHAN (2010)Référence 1 |
Tsang, et. al. (2005)Référence 2 >1000 g P.C. |
LSRO (2002)Référence 3 |
OMSRéférence 4 >1000 g P.C. (stabilisation à terme)Remarque a |
Koletzko, et. al. (2014)Référence 5 |
Min |
Max |
||
Vitamine A |
UI |
250 – 500 |
1200 – 2467 |
538 – 1364 |
680 – 1267 |
583 – 1250 |
1217 – 3333 |
|||
mcg RE |
-- |
[360 – 740] |
[162 – 410] |
[204 – 380] |
[175 – 375] |
[365 – 1000] |
||||
Vitamine D |
UI |
40 – 100 |
800 – 1000Remarque f par jour |
115 – 364 |
75 – 270 |
[400 – 800]Remarque f par jour |
100 – 350 provenant seulement du lait |
|||
Vitamine E |
UI |
0.6 |
[3,0 – 15] |
4,6 – 10,9 |
[3,0 – 12] |
[5,0 – 10,0] |
[3,0 – 15] |
|||
mg α-TE |
-- |
2 – 10 |
[3,1 – 7,3] |
2 – 8 Remarque g |
[3,4 – 6,7] |
2 – 10 |
||||
Vitamine K |
mcg |
8 |
4 – 25 |
6,2 – 9,1 |
4 – 25 |
[6.7 – 8.3] |
4 – 25 |
|||
Vitamine C |
mg |
8 |
10 – 42 |
13,8 – 21,8 |
8,3 – 37 |
[5 – 8] |
18 – 50 |
|||
Thiamine |
mcg |
40 |
125 – 275 |
138 – 218 |
30 – 250 |
[33 – 42] |
127 – 273 |
|||
Riboflavine |
mcg |
60 |
180 – 365 |
192 – 327 |
80 – 620 |
[300 – 383] |
181 – 364 |
|||
Vitamine B6 |
mcg |
35 |
41 – 273 |
115 – 191 |
30 – 250 |
[15]Remarque h |
45 – 273 |
|||
Vitamine B12 |
mcg |
0,15 |
0,08 – 0,7 |
0,23 – 0,27 |
0,08 – 0,7 |
[0,15]Remarque f |
0,09 – 0,73 |
|||
Niacine |
mcg |
250 |
345 – 5000 |
2800 – 4400 |
550 – 5000 |
[720] |
900 – 5000 |
|||
Acide pantothénique |
mcg |
300 |
300 – 1900 |
900 – 1500 |
300 – 1900 |
[667 – 1083] |
450 – 1900 |
|||
Acide folique |
mcg |
4 |
32 – 90 |
19 – 45 |
30 – 45 |
[50]Remarque f |
32 – 91 |
|||
Biotine |
mcg |
2 |
1,5 – 15 |
2,8 – 5,5 |
1,0 – 37 |
[1,3] |
1.5 – 15 |
|||
Choline |
mg |
12 |
7 – 50 |
11,1 – 25,5 |
7 – 23 |
AP |
7,3 – 50 |
|||
Inositol |
mg |
AP |
4 – 48 |
25 – 74 |
4 – 44 |
AP |
4 – 48 |
|||
Taurine |
mg |
AP |
AP |
3,5 – 8,2 |
5 – 12 |
AP |
AP |
|||
Carnitine |
mg |
AP |
AP |
2,2 – 2,6 |
2 – 5,9 |
AP |
AP |
|||
Calcium (Ca) |
mg |
50 |
110 – 130 |
77 – 200 |
123 – 185 |
[134 – 200] |
109 – 182 |
|||
Phosphore(P) |
mg |
25 |
55 – 80 |
46 – 127 |
82 – 109 |
[65 – 98] |
55 – 127 |
|||
Ca:P |
-- |
1,2 – 2 |
AP |
AP |
1,7 – 2,0:1 |
AP |
AP |
|||
Magnésium |
mg |
6 |
7,5 – 13,6 |
AP |
6,8 – 17 |
[4,1 – 8,1] |
7,5 – 13.6 |
|||
Sodium |
mg |
20 – 60 |
63 – 105 |
53 – 105 |
39 – 63 |
[48 – 77] |
63 – 105 |
|||
Potassium |
mg |
80 – 200 |
60 – 120 |
60 – 106 |
60 – 160 |
[81 – 114] |
71 – 177 |
|||
Chlorure |
mg |
55 – 150 |
95 – 161 |
82 – 226 |
60 – 160 |
[74 – 118] |
95 – 161 |
|||
Fer |
mg |
0.15 |
1,8 – 2,7 |
1,538 – 3,636 |
1,7 – 3,0 |
[1.7 – 2.5] |
1,8 – 2,7 |
|||
Zinc |
mg |
0.5 |
1,0 – 1,8 |
0,769 – 2,727 |
1,1 – 1,5 |
[0,42 – 0,67] |
1,3 – 2,3 |
|||
Cuivre |
mcg |
60 |
90 – 120 |
92 – 136 |
100 – 250 |
[58 – 101] |
90 – 210 |
|||
Manganèse |
mcg |
5 |
6,3 – 25 |
0,5 – 6,8 |
6,3 – 25 |
[0,5 – 0,9] |
0,9 – 13,6 |
|||
Iode |
mcg |
5 |
10 – 50 |
7,7 – 54,5 |
6 – 35 |
[26 – 53] |
9 – 50 |
|||
Sélénium |
mcg |
AP |
4,5 – 9 |
1,0 – 4,1 |
1,8 – 5,0 |
[2,6 – 3,9] |
4,5 – 9 | |||
Chrome |
mcg |
AP |
0,027 – 1,12 |
0,08 – 2,05 |
AP |
0,04 – 0,08 |
0,027 – 2,045 |
|||
Molybdène |
mcg |
AP |
0,27 – 4,5 |
0,23 – 0,27 |
AP |
0,16 – 0,32 |
0,27 – 4,5 |
|||
Fluorure |
mcg |
AP |
1,4 – 55 |
NS |
0 – 25 |
AP |
1,4 – 55 |
|||
- NUCLÉOTIDES
Éléments nutritifs |
Exigences du RAD |
Recommandations des experts internationaux |
Profil nutritionnel |
Spécification |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
Nom |
Unité |
ESPGHAN (2010)Référence 1 |
Tsang, et. al. (2005)Référence 2 >1000 g P.C. |
LSRO (2002)Référence 3 |
OMSRéférence 4 >1000 g P.C. (stabilisation à terme)Remarque a |
Koletzko, et. al. (2014)Référence 5 |
Min |
Max |
||
Nucléotides totaux |
mg |
AP |
AP |
AP |
AP |
AP |
AP |
|||
AMP |
mg |
AP |
AP |
0,27 – 0,73 |
AP |
AP |
AP |
|||
CMP |
mg |
AP |
AP |
1,6 – 3,7 |
AP |
AP |
AP |
|||
GMP |
mg |
AP |
AP |
0,03 – 0,54 |
AP |
AP |
AP |
|||
UMP |
mg |
AP |
AP |
0,69 – 0,9 |
AP |
AP |
AP |
|||
Abréviations
- AA
- acide arachidonique
- AAL
- acide alpha-linolénique;
- ADH
- acide docosahexaénoïque
- AGPI
- acides gras polyinsaturés
- AL
- acide linoléique
- AMP
- adénosine monophosphate
- AP
- aucune précision
- Ca:P
- calcium/phosphore
- CMP
- cytosine monophosphate
- Eα-T
- équivalents α-tocophérol
- ER
- équivalents rétinol
- g
- gramme
- GMP
- guanosine monophosphate
- LSRO
- Life Sciences Research Office
- Max.
- maximum
- mcg
- microgramme
- mg
- milligramme
- Min.
- minimum
- NS
- non spécifié
- OMS
- Organisation mondiale de la Santé
- P.C.
- poids corporel
- UMP
- uridine monophosphate
Référence
- Référence 1
-
Agostoni, C., G. Buonocore, V.P. Carnielli, M. De Curtis, D. Darmaun, T. Decsi et coll. « Enteral nutrient supply for preterm infants: commentary from the European Society of Paediatric Gastroenterology, Hepatology and Nutrition Committee on Nutrition », Journal of Pediatric Gastroenterology and Nutrition, vol. 50, p. 85-91, 2010.
- Référence 2
-
Tsang, R.C., R. Uauy, B. Koletzko et S. Zlotkin. Nutrition of the preterm infant: Scientific basis and practical guidelines, Cincinnati (Ohio), Digital Educational Publishing, Inc., 2005.
- Référence 3
-
Klein, C.J. « Nutrient requirements for preterm infant formulas », Journal of Nutrition, vol. 132, p. 1395S-1577S, 2002.
- Référence 4
-
Tudehope, D., et coll. « Nutritional Needs of the Micropreterm Infant », Journal of Pediatrics, vol. 162, no 3, suppl., p. S72-S80, mars 2013.
- Référence 5
-
Koletzko, B., B. Poindexter et R. Uauy. Nutritional Care of Preterm Infants: Scientific Basis and Practical Guidelines, Karger, p. 99-120, 2014.
Remarque
- Remarque a
-
Calculé sur la base de la moyenne des recommandations énergétiques maximales et minimales pour la population indiquée; 120 kcal/kg/j pour une stabilisation à terme.
- Remarque b
-
3,6-4,1 g/100 kcal pour les nourrissons dont le poids corporel est inférieur à 1 kg; 3,2-3,6 g/100 kcal pour les nourrissons dont le poids corporel se situe entre 1 et 1,8 kg.
- Remarque c
-
Acides gras totaux : 8 % (minimum) et 25 % (maximum)
- Remarque d
-
La recommandation minimale pour l’AA n’a pas été établie. Maximum : 0,6 % des acides gras totaux, avec la condition supplémentaire que le ratio AA:ADH se situe dans la plage 1,5-2,0:1.
- Remarque e
-
La recommandation minimale pour l’ADH n’a pas été établie. Maximum : 0,35 % des acides gras totaux, avec la condition supplémentaire que le ratio AA:ADH se situe dans la plage 1,5-2,0:1.
- Remarque f
-
Recommandation donnée par jour, indépendamment du poids corporel et de l’apport énergétique.
- Remarque g
-
Le ratio vitamine E:AGPI (mg d’équivalents α-tocophérol par g d’AGPI) devrait être supérieur à 1,5 mg/g α-tocophérol par g d’AGPI).
- Remarque h
-
Recommandation par gramme de protéines consommé.
Appendice 4. Résumé des fluides et des apports nutritionnels recommandés par les comités d’experts internationaux
| Comité d’experts | Plage d’apport liquidien (ml/kg/jour) | Plage d’apport énergétique (kcal/kg/jour) | Poids du nourrisson (g) |
| Koletzko (2014)Référence 1 | 135-200 | 110-130 | 1000-1500 |
| ESPGHAN (2010)Référence 2 | 135-200 | 110-135 | 1000-1800 |
| OMS (2006)Référence 3 | S. O. | 105-135 | > 1000 |
| Tsang (2005)Référence 4 et AAP (2014)Référence 5 | 135-190 | 110-130* | 1000-1500 |
| LSRO (2002)Référence 6 | 150 | 110-135 | 1000 |
Références
- Référence 1
-
Nutritional Care of Preterm Infants: Scientific Basis and Practical Guidelines. Koletzko B, Poindexter B, Uauy R (eds). Basel, Switzerland: Karger, 2014:99-120.
- Référence 2
-
Agostoni C, Buonocore G, Carnielli VP, De Curtis M, Darmaun D, Decsi T, et al. Enteral nutrient supply for preterm infants: commentary from the European Society of Paediatric Gastroenterology, Hepatology, and Nutrition Committee on Nutrition. J Pediatr Gastroenterol Nutr 2010;50:85-91.
- Référence 3
-
Tudehope D. et al. Nutritional Needs of the Micropreterm Infant March 2013, Volume 162, Issue 3, Supplement, Pages S72–S80.
- Référence 4
-
Nutrition of the Preterm Infant: Scientific Basis and Practical Guidelines. 2nded. Tsang RC, Uauy R, Koletzko B, Zlotkin SH (eds). Cincinnati, OH: Digital Educational Publishing Inc, 2005: 23-44, 201-244.
- Référence 5
-
American Academy of Pediatrics Committee on Nutrition. Nutritional needs of preterm infant. In: Kleinman RE, Greer FR (eds). Pediatric Nutrition. American Academy of Pediatrics, 2014:113-162.
- Référence 6
-
Klein CJ. Nutrient requirements for preterm infant formulas. J Nutr 2002;132:1395S-577S
* Il est à noter que pour les prématurés atteints d’une maladie pulmonaire chronique, l'apport énergétique recommandé est augmenté à 140 kcal/kg/jour, et pour les prématurés de poids extrêmement faible à la naissance (PEFN), l'apport énergétique maximal recommandé est de 150 kcal/kg/jour. Dans des situations cliniques particulières, les apports peuvent atteindre 160 à 180 kcal/kg/jour (AAP, 2014).
Appendice 5. Composition du lait maternel pour prématurés
Note : Ces données sont reommandées pour remplir le modèle ci-dessus, ainsi que pour normaliser l'approche.
Éléments nutritifs |
Unités |
Lait humain pour prématurés |
Lait humain pour prématurés |
|---|---|---|---|
MACRONUTRIMENTS |
|||
Protéines Référence 1Remarque a |
g |
2,1 |
1,5 |
Lipides Référence 1Remarque a |
g |
4,9 |
3,5 |
- AL Référence 2 |
g |
0,72 |
0,48 |
- AAL Référence 2 |
g |
0,045 |
0,03 |
- AL:AAL |
-- |
16:1 |
16:1 |
- AA Référence 2 |
g |
0,029 |
0,017 |
- ADH Référence 2 |
g |
0,017 |
0,011 |
Glucides Référence 2 |
g |
10,9 |
7,3 |
VITAMINES ET MINÉRAUX |
|||
Vitamine A Référence 2 |
IU |
72 |
48 |
Vitamine D Référence 2 |
IU |
12 |
8 |
Vitamine E Référence 2 |
IU |
0,59 |
0,39 |
Vitamine K Référence 2 |
mcg |
3 |
2 |
Vitamine C Référence 2 |
mg |
6,6 |
4,4 |
Thiamine Référence 2 |
mcg |
13,35 |
8,9 |
Riboflavine Référence 2 |
mcg |
40,5 |
27 |
Vitamine B6 Référence 2 |
mcg |
9,3 |
6,2 |
Vitamine B12 Référence 2 |
mcg |
0,03 |
0,02 |
Niacine Référence 2 |
mcg |
315 |
210 |
Acide pantothénique Référence 2 |
mcg |
345 |
230 |
Acide folique Référence 2 |
mcg |
4,65 |
3,1 |
Biotine Référence 2 |
mcg |
0,81 |
0,54 |
Choline Référence 3 |
mg |
23,7 |
15,8 |
Inositol Référence 4 |
mg |
29,1 |
19,4 |
VITAMINES ET MINÉRAUX (suite) |
|||
Taurine Référence 2 |
mg |
6 |
4 |
Carnitine Référence 2 |
mg |
1,05 |
0,7 |
Calcium Référence 1Remarque a |
mg |
35 |
25 |
Phosphore Référence 1Remarque a |
mg |
21 |
15 |
Ca:P |
mg-mg |
1.67: 1 |
1,67: 1 |
Magnésium Référence 2 |
mg |
4,95 |
3,3 |
Sodium Référence 2 |
mg |
42 |
28 |
Potassium Référence 2 |
mg |
75 |
50 |
Chlorure Référence 2 |
mg |
87 |
58 |
Fer Référence 2 |
mg |
0,135 |
0,09 |
Zinc Référence 2 |
mg |
0,56 |
0,37 |
Cuivre Référence 2 |
mcg |
57 |
38 |
Manganèse Référence 2 |
mcg |
0,54 |
0,36 |
Iode Référence 2 |
mcg |
26,7 |
17,8 |
Sélénium Référence 2 |
mcg |
3,6 |
2,4 |
Chrome Référence 5Remarque b |
mcg |
0,038 |
0,025 |
Molybdène Référence 5 |
mcg |
0,3 |
0,2 |
Fluorure Remarque c |
mcg |
NS |
NS |
NUCLÉOTIDES |
|||
Nucléotides totaux |
mg |
NS |
NS |
AMP |
mg |
NS |
NS |
CMP |
mg |
NS |
NS |
GMP |
mg |
NS |
NS |
UMP |
mg |
NS |
NS |
Abréviations
- AAL
- Acide alpha-linolénique
- AA
- Acide arachidonique
- ADH
- Acide docosahexaénoïque
- AL
- Acide linoléique
- AMP
- Adénosine monophosphate
- Ca:P
- Rapport calcium/phosphore
- CMP
- Cytosine monophosphate
- g
- Gramme
- GMP
- Guanosine monophosphate
- mcg
- Microgramme
- mg
- Milligramme
- NS
- Non spécifié
- OMS
- Organisation mondiale de la Santé
- P.C.
- Poids corporel
- UMP
- Uridine monophosphate
Références
- Référence 1
-
Gidrewicz DA, Fenton TR. « A systematic review and meta-analysis of the nutrient content of preterm and term breast milk ». BMC Pediatrics, vol. 14, p. 216, 30 août 2014.
- Référence 2
-
Koletzko B, Poindexter B, Uauy R. Nutrtional Care of Preterm Infants. Scientific basis and practical guidelines. Bâle (Suisse), Karger AG, 2014.
- Référence 3
-
Maas C, Franz AR, Shunova A, Mathes M, Bleeker C, Poets CF, Schleicher E, Bernhard W. « Choline and polyunsaturated fatty acids in preterm infants' maternal milk ». European Journal of Nutrition, vol. 56, no 4, p. 1733-1742, juin 2017.
- Référence 4
-
Moles L, Manzano S, Fernández L, Montilla A, Corzo N, Ares S, Rodríguez JM, Espinosa-Martos I. « Bacteriological, biochemical, and immunological properties of colostrum and mature milk from mothers of extremely preterm infants ». Journal of Pediatric Gastroenterology and Nutrition, vol. 60, no 1, p. 120-126, janvier 2015.
- Référence 5
-
Klein CJ. « Nutrient requirements for preterm infant formulas ». Journal of Nutrition, vol. 132, no 6, suppl. 1, p. 1395S-1577S, juin 2002.
Remarques
- Pour la référence 1, 71 kcal/100 mL; pour les autres, ~67 kcal/100 mL
- Remarque a
-
Les valeurs relatives aux protéines, aux lipides, au calcium et au phosphore sont basées sur les données de la semaine 2 (méta-analyse); toutes les autres valeurs sont basées sur les données obtenues durant les semaines 1 à 4
- Remarque b
-
Les valeurs pour le chrome sont basées sur le lait humain pour nourrissons né à terme (valeurs utilisées par le LSRO, 2002)
- Remarque c
-
Non Spécifié en raison des fortes fluctuations (dues vraisemblablement à la variation des taux de fluorure dans l'eau)