
Archived - Molecular phylogenetic used to identify HCV transmission

 Download this article as a PDF
Download this article as a PDFPublished by: The Public Health Agency of Canada
Issue: Volume 45–9: Outbreaks
Date published: September 5, 2019
ISSN: 1481-8531
Submit a manuscript
About CCDR
Browse
Volume 45–9, September 5, 2019: Outbreaks
Surveillance
Molecular surveillance of hepatitis C virus genotypes identifies the emergence of a genotype 4d lineage among men in Quebec, 2001–2017
DG Murphy1, R Dion1,2, M Simard3, ML Vachon4, V Martel-Laferrière5, B Serhir1, J Longtin1
Affiliations
1 Institut national de santé publique du Québec, Laboratoire de santé publique du Québec, Sainte-Anne-de-Bellevue, QC
2 École de santé publique de l’Université de Montréal, Département de médecine sociale et préventive, Montréal, QC
3 Institut national de santé publique du Québec, Bureau d’information et d’études en santé des populations, Québec, QC
4 Centre hospitalier de l’Université Laval, Québec, QC
5 Centre de recherche du Centre hospitalier de l'Université de Montréal, Montréal, QC
Correspondence
Suggested citation
Murphy DG, Dion R, Simard M, Vachon ML, Martel-Laferrière V, Serhir B, Longtin J. Molecular surveillance of hepatitis C virus genotypes identifies the emergence of a genotype 4d lineage among men in Quebec, 2001–2017. Can Commun Dis Rep 2019;45(9):230–7. https://doi.org/10.14745/ccdr.v45i09a02
Keywords: HCV, genotype, G4d, surveillance, phylogenetic analyses, cluster, molecular epidemiology, men who have sex with men, MSM
Abstract
Background: Molecular phylogenetics are generally used to confirm hepatitis C virus (HCV) transmission events. In addition, the Laboratoire de santé publique du Québec (LSPQ) has been using molecular phylogenetics for surveillance of HCV genotyping since November 2001.
Objectives: To describe the emergence of a specific lineage of HCV genotype 4d (G4d) and its characteristics using molecular phylogenetics as a surveillance tool for identifying HCV strain clustering.
Methods: The LSPQ prospectively applied Sanger sequencing and phylogenetic analysis to determine the HCV genotype on samples collected from November 2001 to December 2017. When a major G4d cluster was identified, demographic information, HIV-infection status and syphilis test results were analyzed.
Results: Phylogenetic analyses performed on approximately 22,000 cases identified 122 G4d cases. One major G4d cluster composed of 37 cases was singled out. Two cases were identified in 2010, 10 from 2011–2014 and 25 from 2015–2017. Cases in the cluster were concentrated in two urban health regions. Compared to the other G4d cases, cluster cases were all male (p<0.001) and more likely to be HIV-positive (adjusted risk ratio: 4.4; 95% confidence interval: 2.5–7.9). A positive syphilis test result was observed for 27 (73%) of the cluster cases. The sequences in this cluster and of four outlier cases were located on the same monophyletic lineage as G4d sequences reported in HIV-positive men who have sex with men (MSM) in Europe.
Conclusion: Molecular phylogenetics enabled the identification and surveillance of ongoing transmission of a specific HCV G4d lineage in HIV-positive and HIV-negative men in Quebec and its cross-continental spread. This information can orient intervention strategies to avoid transmission of HCV in MSM.
Introduction
Hepatitis C virus (HCV) affects 70 million people worldwide and is a major public health concern. In Canada, nearly 250,000 persons, or 0.7% of the population, are estimated to be chronically infected with HCV and up to 44% of this population may be unaware of their statusFootnote 1. In Quebec, 1,027 cases of HCV were reported in 2017 (incidence rate [IR] of newly reported cases of 12.2 per 100,000 population), with a projection estimate of 1,312 cases (IR of 15.5 per 100,000 population) for 2018Footnote 2.
Based on prospective cohorts such as SurvUDI network and the Engage study, people who inject drugs and men who have sex with men (MSM) are disproportionally affected by HCV infection compared to the general populationFootnote 2Footnote 3. Chronic HCV infection, if untreated, can result in fibrosis, cirrhosis, liver failure and hepatocellular carcinoma. Early identification of HCV infection followed by treatment is crucial in reducing HCV transmission, associated morbidity and mortality and health care costsFootnote 4.
HCV is mostly transmitted by the parenteral route, the most common risk factors being injection drug use and blood or blood product transfusion (prior to the introduction of blood donor screening). Sexual transmission of HCV is less common, but possible. Recent data indicates an increase in HCV prevalence among MSM, especially among those coinfected with HIVFootnote 5. High-risk behaviours, including unprotected sex, have been identified as determinants for HCV transmission. Ulcerative sexually transmitted infections, such as syphilis, has also been associated with increased risk of HCV acquisition in MSMFootnote 6Footnote 7.
HCV is currently classified into eight genotypesFootnote 8. Genotypes are further divided into subtypes, of which 89 have been confirmedFootnote 8. At the Laboratoire de santé publique du Québec (LSPQ), Institut national de santé publique du Québec, HCV genotyping has been performed routinely since November 2001 for patient management, as genotyping helps to guide antiviral treatment of chronic infection.
Genotyping performed by sequencing analysis not only informs optimal treatment choice, it can also be a powerful molecular surveillance tool for identification of circulating virus strains. Although molecular phylogenetics cannot determine the time of infection acquisition, it can detect transmission clusters. The laboratory can then notify public health authorities that a particular HCV strain is spreading in the populationFootnote 9Footnote 10.
Molecular phylogenetics has not been used extensively for ongoing prospective surveillance, but studies have shown it to be a potentially useful tool. For example, nucleic acid sequencing and phylogenetic analysis have been used to identify and confirm HCV transmission events in the health care setting and to characterize transmission dynamics in the communityFootnote 11Footnote 12Footnote 13Footnote 14Footnote 15Footnote 16Footnote 17Footnote 18. Phylogenetic strain clustering has also been used to investigate HCV transmission networks among HIV-infected MSMFootnote 19Footnote 20. A recent study supported the feasibility of applying nucleic acid sequencing and phylogenetics to identify recent HCV transmission clusters in individuals with unknown histories of transmissionFootnote 21.
The objective of this article is to describe the use of molecular sequencing and phylogenetics as a tool for prospective surveillance of HCV transmission at the population level. Namely, we show how this approach identified the emergence and ongoing transmission of HCV genotype 4d (G4d) among HIV-positive and HIV-negative men in Quebec and how we were able to identify its source.
Methods
Study population
In Quebec, HCV genotyping is conducted at the LSPQ. Samples submitted for routine HCV genotyping from November 13, 2001 to December 31, 2017 were included in this study. Samples were submitted from hospital laboratories and public or private clinics in Quebec. Data on the G4d cases originated from the LSPQ’s laboratory information system and included basic demographic information (age, sex, health region of residence), date of specimen collection and test results for HIV and syphilis infections. These were retrieved from the information system using patients’ unique identifier numbers. Data on exposures or risk factors were not collected. Genotype results are not routinely reported to the public health regional authorities.
Genotyping and nucleotide sequence analysis
The HCV genotype of each sample was assessed as it was received. Most samples were received within 30 days of blood collection. Viral ribonucleic acid (RNA) extraction from serum or plasma as well as reverse transcription polymerase chain reaction (RT-PCR), DNA sequencing and genotyping based on nonstructural protein 5B (NS5B) sequences were performed as previously describedFootnote 22. Quebec’s G4d sequences were compared to sequences previously reported in HIV-infected MSM in the Netherlands and FranceFootnote 23Footnote 24Footnote 25. To compare Quebec’s G4d sequences with those reported in the van de Laar et al. studyFootnote 20 for participants from England, France, Germany and the Netherlands, the corresponding NS5B nucleotide sequence was obtained for a subset of Quebec’s G4d isolates by use of reverse primer DM503 (5′-CCACGCTCTCAACGGTGGTAC-3) and forward primer DM101 to generate a 805 base pair sequence fragmentFootnote 22. These were selected on the basis of sample availability. Phylogenies were estimated by the neighbour-joining method according to the maximum composite likelihood model of nucleotide substitution implemented in Molecular Evolutionary Genetic Analysis (MEGA) software version 6Footnote 26. As many as 1,000 bootstrap replicates were performed to evaluate the robustness of the phylogeny. Sequences were considered to be part of a cluster if the following three criteria were met: showed less than 3% nucleotide difference; displayed a bootstrap support value of greater than 70%; and consisted of at least three cases. The closeness of cases in terms of time and space was not included in this definition. G4d sequences for isolates reported in this study were submitted to GenBank and can be retrieved under accession numbers MK950000 to MK950149. NS5B sequences for isolates 3_QC55 (EF116095), 10_QC307 (EF116147), and 27_QC382 (FJ462437) were submitted previously to GenBank.
Statistical analysis
Descriptive analysis of the data was conducted using Epi Info software version 7.2.2.6Footnote 27 and statistical package SAS version 9.3Footnote 28. This analysis was based on specimen collection dates, grouped as quarterly periods. For univariate analysis, statistical comparisons of categorical variables (age group, sex, health region, G4d HCV cluster and HIV infection status) were performed using bilateral Mantel–Haenszel chi square test and modified Poisson regression with a robust error variance sandwich estimationFootnote 29. To assess their link with cluster A outcome, the multivariate analysis using robust Poisson regression included the following independent variables in the model: HIV infection (yes versus no or unknown) and age group (50 years or older vs younger than 50 years). Given that none of the cluster A cases were female, this analysis was restricted to males. Statistically significant levels for confidence intervals (CI) of risk ratio (RR) estimates and bilateral p values were set at 5%.
Results
From November 13, 2001 to December 31, 2017, HCV genotypes were determined from a total of approximately 22,000 cases. G1 was the most prevalent genotype (59.7%), followed by G3 (25.7%), G2 (8.6%), G4 (3.8%), G6 (1.6%), G5 (0.6%) and G7 (0.01% [three cases only]). Phylogenetic analyses of NS5B sequences revealed notable clusters mainly among genotypes 1a, 1b, 2b, 3a and 4d. One major G4d cluster composed of 37 cases was singled out and is described in this study.
Overall, G4 was found in 834 cases. Five G4 cases displayed low levels of viremia and the genotype was determined based on the 5′UTR (5′ untranslated region) sequence, which is too conserved for subtype determination. G4d was found in 122 (14.7%) of 829 G4 cases for which the subtype was available.
Strain clustering of G4d cases
The first G4d case was recorded in December 2001 (Figure 1). From 2002 to 2014, an average of six cases per year was observed. In 2015, 16 cases were noted. This increase continued throughout 2016 and 2017. A phylogenetic tree was constructed based on NS5B sequences for the 122 G4d cases and analyzed for strain clustering. Four G4d clusters (named A to D) were identified (Figure 1 and Figure 2). Cluster A comprised 37 cases, clusters B and C each comprised four cases, and cluster D comprised three cases. G4d cluster A cases first appeared in the second quarter of 2010. Cases within each of clusters B, C and D were scattered in time (Figure 1). An increase in cluster A cases was observed from the second quarter of 2015 to the fourth quarter of 2017, for which 24 of 32 (75%) G4d cases were part of this cluster.
Figure 1: Number of hepatitis C virus genotype 4d cases by year and quarter of specimen collection and sequence clusters, November 2001 to December 2017, Quebec, Canada

Text description: Figure 1
Figure 1: Number of hepatitis C virus genotype 4d cases by year and quarter of specimen collection and sequence clusters, November 2001 to December 2017, Quebec, Canada
| Year and quarter of specimen collection | A | B | C | D | Outliers to A | None |
|---|---|---|---|---|---|---|
| 2001-4 | 0 | 0 | 0 | 0 | 0 | 1 |
| 2002-1 | 0 | 0 | 0 | 0 | 0 | 1 |
| 2002-2 | 0 | 0 | 0 | 0 | 0 | 3 |
| 2002-3 | 0 | 0 | 0 | 0 | 0 | 1 |
| 2002-4 | 0 | 0 | 0 | 0 | 0 | 1 |
| 2003-1 | 0 | 0 | 0 | 0 | 0 | 6 |
| 2003-2 | - | - | - | - | - | - |
| 2003-3 | 0 | 0 | 0 | 0 | 0 | 4 |
| 2003-4 | 0 | 0 | 1 | 0 | 0 | 2 |
| 2004-1 | 0 | 0 | 0 | 0 | 0 | 4 |
| 2004-2 | 0 | 0 | 1 | 0 | 0 | 2 |
| 2004-3 | 0 | 0 | 0 | 0 | 0 | 2 |
| 2004-4 | - | - | - | - | - | - |
| 2005-1 | 0 | 0 | 1 | 0 | 0 | 0 |
| 2005-2 | - | - | - | - | - | - |
| 2005-3 | 0 | 0 | 0 | 0 | 0 | 1 |
| 2005-4 | - | - | - | - | - | - |
| 2006-1 | 0 | 0 | 0 | 0 | 0 | 2 |
| 2006-2 | 0 | 0 | 0 | 0 | 0 | 1 |
| 2006-3 | 0 | 0 | 0 | 0 | 0 | 2 |
| 2006-4 | - | - | - | - | - | - |
| 2007-1 | 0 | 1 | 0 | 0 | 0 | 1 |
| 2007-2 | - | - | - | - | - | - |
| 2007-3 | 0 | 1 | 0 | 0 | 0 | 0 |
| 2007-4 | 0 | 0 | 0 | 0 | 0 | 3 |
| 2008-1 | 0 | 0 | 0 | 0 | 0 | 1 |
| 2008-2 | 0 | 0 | 0 | 0 | 0 | 1 |
| 2008-3 | - | - | - | - | - | - |
| 2008-4 | 0 | 1 | 1 | 0 | 0 | 0 |
| 2009-1 | 0 | 0 | 0 | 0 | 0 | 1 |
| 2009-2 | - | - | - | - | - | - |
| 2009-3 | - | - | - | - | - | - |
| 2009-4 | 0 | 0 | 0 | 0 | 0 | 1 |
| 2010-1 | 0 | 0 | 0 | 0 | 0 | 1 |
| 2010-2 | 1 | 0 | 0 | 1 | 0 | 0 |
| 2010-3 | 1 | 1 | 0 | 0 | 0 | 0 |
| 2010-4 | 0 | 0 | 0 | 0 | 0 | 3 |
| 2011-1 | 1 | 0 | 0 | 1 | 0 | 3 |
| 2011-2 | - | - | - | - | - | - |
| 2011-3 | 1 | 0 | 0 | 0 | 0 | 1 |
| 2011-4 | 1 | 0 | 0 | 0 | 0 | 0 |
| 2012-1 | 2 | 0 | 0 | 0 | 0 | 0 |
| 2012-2 | 1 | 0 | 0 | 0 | 1 | 0 |
| 2012-3 | 0 | 0 | 0 | 0 | 0 | 4 |
| 2012-4 | 0 | 0 | 0 | 0 | 1 | 0 |
| 2013-1 | 0 | 0 | 0 | 0 | 0 | 1 |
| 2013-2 | 0 | 0 | 0 | 0 | 0 | 2 |
| 2013-3 | 2 | 0 | 0 | 1 | 0 | 1 |
| 2013-4 | - | - | - | - | - | - |
| 2014-1 | 0 | 0 | 0 | 0 | 0 | 1 |
| 2014-2 | 2 | 0 | 0 | 0 | 0 | 0 |
| 2014-3 | 0 | 0 | 0 | 0 | 0 | 1 |
| 2014-4 | 0 | 0 | 0 | 0 | 0 | 1 |
| 2015-1 | 1 | 0 | 0 | 0 | 1 | 3 |
| 2015-2 | 5 | 0 | 0 | 0 | 0 | 0 |
| 2015-3 | 5 | 0 | 0 | 0 | 0 | 0 |
| 2015-4 | 0 | 0 | 0 | 0 | 0 | 1 |
| 2016-1 | 2 | 0 | 0 | 0 | 0 | 0 |
| 2016-2 | 1 | 0 | 0 | 0 | 0 | 0 |
| 2016-3 | 2 | 0 | 0 | 0 | 1 | 2 |
| 2016-4 | 1 | 0 | 0 | 0 | 0 | 0 |
| 2017-1 | 1 | 0 | 0 | 0 | 0 | 0 |
| 2017-2 | 2 | 0 | 0 | 0 | 0 | 2 |
| 2017-3 | 4 | 0 | 0 | 0 | 0 | 2 |
| 2017-4 | 1 | 0 | 0 | 0 | 0 | 0 |
Figure 2: Phylogenetic tree of genotype 4d NS5B hepatitis C virus isolate sequences, Quebec, Canada, November 2001 to December 2017 (n=122)
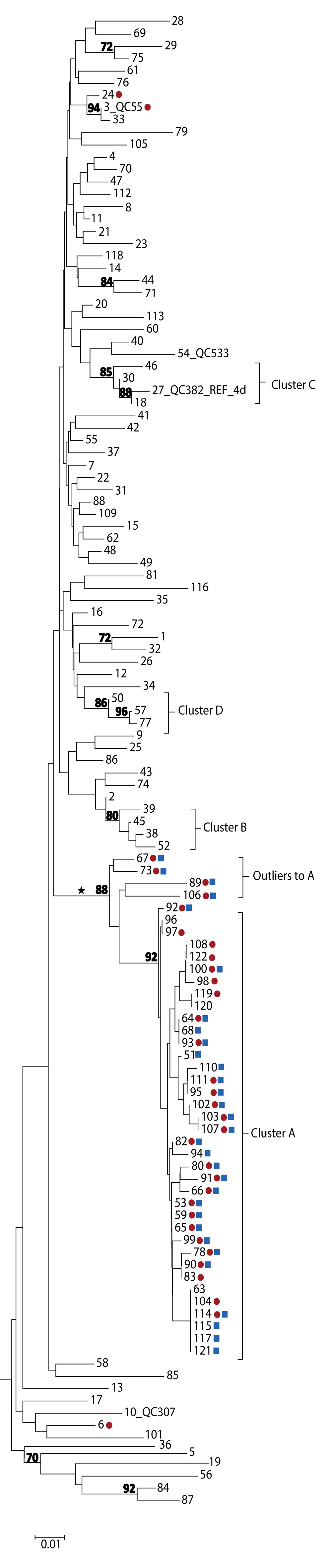
Text description: Figure 2
Figure 2: Phylogenetic tree of genotype 4d NS5B hepatitis C virus isolate sequences, Quebec, Canada, November 2001 to December 2017 (n=122)
The phylogenetic tree shows the genetic relatedness of different hepatitis C virus (HCV) genotype 4d non-structural protein 5B sequences (nucleotide positions 8276-8615) for cases from Quebec, between November 2001 and December 2017. The lengths of the horizontal lines or branches correspond to the genetic distance between sequences. The sequences forming a cluster (named A to D) are identified. The 34 red dots indicate HCV sequences from HIV-1-positive individuals among the 122 G4d cases. The 31 blue squares indicate treponemal test positive cases among the 37 cluster A and four outlier to A cases. The cross-continental monophyletic lineage is indicated by an asterisk. The numbers at nodes represent the percentage bootstrap values (only values greater than 70% are shown on nodes of non-identical sequences).
Four cases (designated as outliers to cluster A) that localized on the same monophyletic lineage as cluster A cases were also identified. However, these were excluded as they displayed greater than 3% nucleotide differences from cluster A strains (Figure 1 and Figure 2). Outliers to cluster A cases were identified during the same period as cluster A cases. Sequences of cluster A and of the four outliers localized on the same monophyletic lineage as G4d sequences were reported in HIV-positive MSM in England, France, Germany and the Netherlands (Figures A1 and A2 in Appendix 1)Footnote 20Footnote 23Footnote 24Footnote 25.
Demographics and other sexually transmitted infections
On univariate analysis, cluster A cases, compared to the other G4d cases, were more likely to be male (100% vs 62%; RR: undefined; p<0.001), HIV-positive (73% vs 8%; RR: 6.99; 95% CI: 3.80–12.84; p<0.001), and 50 years or older (60% vs 34%; RR: 2.04; 95% CI: 1.18–3.54; p=0.01). In multivariate analysis restricted to male cases, age group (adjusted RR [aRR]: 1.66; 95% CI: 1.11–2.50; p=0.02) and HIV infection (aRR: 4.43; 95% CI: 2.49–7.88; p<0.001) remained significantly associated with the cluster A outcome. Of cluster A cases, 27 (73%) had a history of positive treponemal test results for syphilis and 20 (57%) were both HIV-positive and had a history of positive treponemal test results.
Cluster A cases resided in seven of the 18 health regions in Quebec, but were concentrated in two nonadjacent and predominantly urban regions. These two regions accounted for 78.3% of cluster A cases (Table 1). Of note, in one of the urban regions (health region Y), 14 of 24 (58.3%) observed G4d cases were part of cluster A. Although the IR of newly notified cases of HCV in health region Y was fourfold lower than in health region X from 2013–2017, an equivalent number of G4d cluster A cases was observed in these two health regions (14 vs 15). The cases not in cluster A resided in 14 health regions, of which the one with the largest population of the province accounted for 49.4% of cases.
| Health region | Population in 2015 (n)Footnote a of Table 1 | Mean annual HCV cases, 2013–2017Footnote 2 | HCV G4d cases from November 2001 to December 2017Footnote b of Table 1 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | % | IRFootnote c of Table 1 | In cluster A | Not in cluster A | Total | |||||
| n | % | n | % | n | % | |||||
| X | 1,992,106 | 399 | 36.6 | 20.0 | 15 | 40.5 | 42 | 49.4 | 57 | 46.7 |
| Y | 736,787 | 92 | 8.4 | 12.5 | 14 | 37.8 | 10 | 11.8 | 24 | 19.7 |
| Others | 5,562,462 | 600 | 55.0 | 10.8 | 8 | 21.6 | 33 | 38.8 | 41 | 33.6 |
| Total | 8,291,355 | 1,091 | 100.0 | 13.2 | 37 | 100.0 | 85 | 100.0 | 122 | 100.0 |
Discussion
In this study, we found that HCV molecular phylogenetics identified the transmission of a specific lineage (cluster A) of HCV G4d among men in Quebec; this would otherwise have remained undocumented. The G4d cluster A strains were found to be concentrated in two urban health regions (X and Y) and localized on the same monophyletic lineage as G4d sequences reported in HIV-positive MSM in England, France, Germany and the NetherlandsFootnote 20Footnote 23Footnote 24Footnote 25 and later in SpainFootnote 30. Although it is likely that this particular G4d cluster A strain was introduced to Canada from Europe, the origin of this strain in Quebec remains unknown.
These findings suggest that high-risk sexual behaviour may be the mode of spread of G4d among the cluster A cases. The international transmission network of this strain in HIV-infected MSM are not injection drug users. There was also a high prevalence of positive treponemal test results. Syphilis is considered as a marker of high-risk sexual behaviourFootnote 31. Ulcerative sexually transmitted infections, such as syphilis, have been associated with increased risk of sexual HCV acquisition in MSMFootnote 6Footnote 7.
In 2015, the emergence of this cluster was brought to the attention of provincial public health authorities and the two regional public health authorities most affected. Since sexually active HIV-negative MSM are also at risk of HCV infection, annual HCV testing is recommended for MSM who engage in HIV preexposure prophylaxisFootnote 32.
Though most cases in cluster A were coinfected with HIV, 27% (10/37) had no laboratory evidence of HIV infection. This indicates likely transmission from HIV-positive to HIV-negative men or vice versa. HCV strain clustering, including G4d, among HIV-positive and HIV-negative MSM has been observed in France and the Netherlands in study participants enrolled in HIV preexposure prophylaxis programsFootnote 24Footnote 33.
Strengths and limitations
The main strength of this study is the high number of HCV-infected cases (about 22,000) for whom uniform sequencing results were available through the study period. An added strength was that the genotyping was performed in the same central reference laboratory. The estimated cumulative number of HCV chronic infections in Quebec reached 42,000 in 2017; nearby 75% can be assumed to be viremicFootnote 2. Thus, the number of individuals included in this study represents a significant proportion of cases who have been diagnosed and are viremic. Nevertheless, G4d cluster A cases are likely to be underestimated as some may not have been diagnosed; in addition, genotyping tests may not have been requested for all cases. Even with this underestimation, the genotyping data was probably unbiased given that it was not directed at specific subgroups of the population.
A limitation of this analysis is that there is no strict definition of strain clustering, as it varies depending on the region of the genome analyzed and the study population. In addition, the rate of HCV evolution can vary over time, based on the individual, and may impact clustering of viral strains. Also, the use of longer HCV sequences could have increased the accuracy of cluster identificationFootnote 34. This study did not include data about determinants, exposures, sexual orientation or epidemiologic links between cases, and could not distinguish the mode of transmission of infection; as such, it can only provide indirect clues of HCV propagation among Quebecois men, most probably MSM.
Conclusion
Molecular phylogenetic-based surveillance revealed an ongoing transmission in Quebec of a specific cluster of HCV G4d. This cluster localized on the same monophyletic lineage as G4d sequences reported in HIV-infected MSM in several countries in Europe, indicating a cross-continental spread of this specific lineage. Phylogenetic analysis results coupled with basic demographic data produces an epidemiologic profile of HCV cases that can orient preventive interventions to avoid HCV transmission among sexually active HIV-positive and HIV-negative MSM.
Authors’ statement
- DGM — Conceptualization, methodology, software, data collection and conservation, validation, official analysis, writing of the first draft, display, supervision, project administration
- RD — Conceptualization, methodology, software, official analysis, writing the first draft, review and revision of the final version, display
- MS — Conceptualization, methodology, software, official analysis, review and revision of the final version
- MLV — Clinical expertise, review and revision of the final version
- VML — Clinical expertise, review and revision of the final version
- BS — Data collection, review and revision of the final version
- JL — Project administration, review and revision of the final version, supervision
Conflict of interest
None.
Acknowledgements
The authors thank L Désautels, J Ménard, M Morin, A Chammat and JL Aguilar for laboratory testing of specimens and collaboration.
Funding
This work was supported by the Laboratoire de santé publique du Québec, Institut national de santé publique du Québec. The work of VML is supported by a Chercheur boursier Junior 1 award from the Fonds de recherche du Québec – Santé.
Appendix: Supplementary data
Figure A1: Phylogenetic tree of genotype 4d NS5B hepatitis C virus isolate sequences: cases in Quebec compared to cases in France and the Netherlands
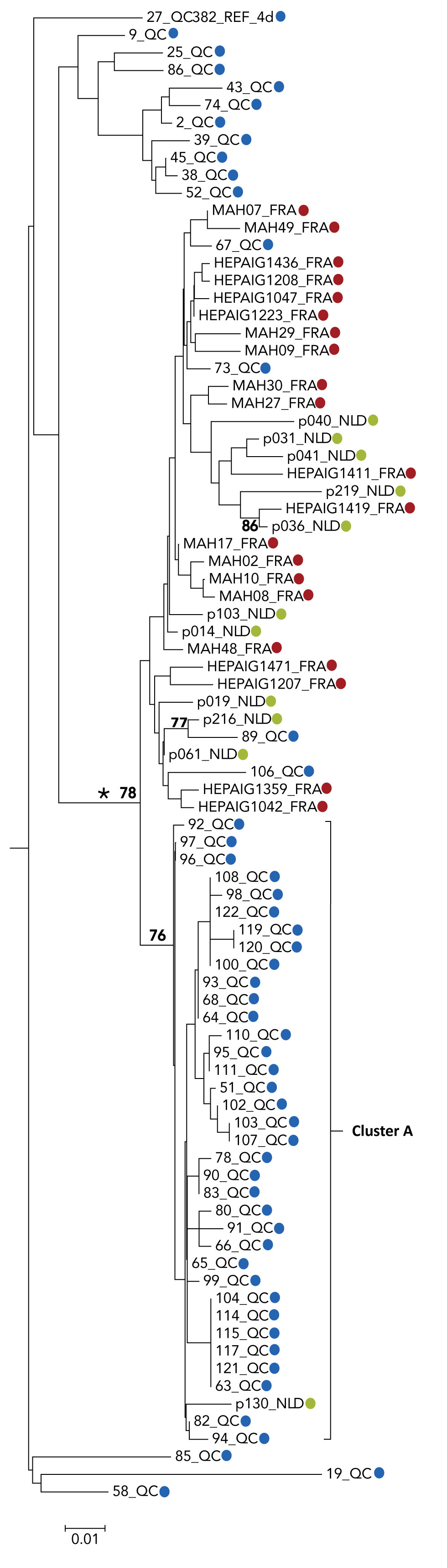
Text description: Figure A1
Figure A1: Phylogenetic tree of genotype 4d NS5B hepatitis C virus isolate sequences: cases in Quebec compared to cases in France and the Netherlands
The phylogenetic tree shows the genetic relatedness of different hepatitis C virus genotype 4d non-structural protein 5B sequences (nucleotide positions 8276-8615) of selected cases from Quebec, France and the Netherlands. The lengths of the horizontal lines or branches correspond to the genetic distance between sequences. The Quebec cluster A is identified. The blue dots represent Quebec, 53 cases, red dots represent France, 21 cases and the green dots represent the Netherlands, 11 cases. The cross-continental monophyletic lineage is indicated by an asterisk. The numbers at nodes represent the percentage bootstrap values (only values greater than 70% are shown on nodes of non-identical sequences).
Figure A2: Phylogenetic tree of genotype 4d NS5B hepatitis C virus isolate sequences: cases in Quebec compared to cases in England, France, Germany and the Netherlands
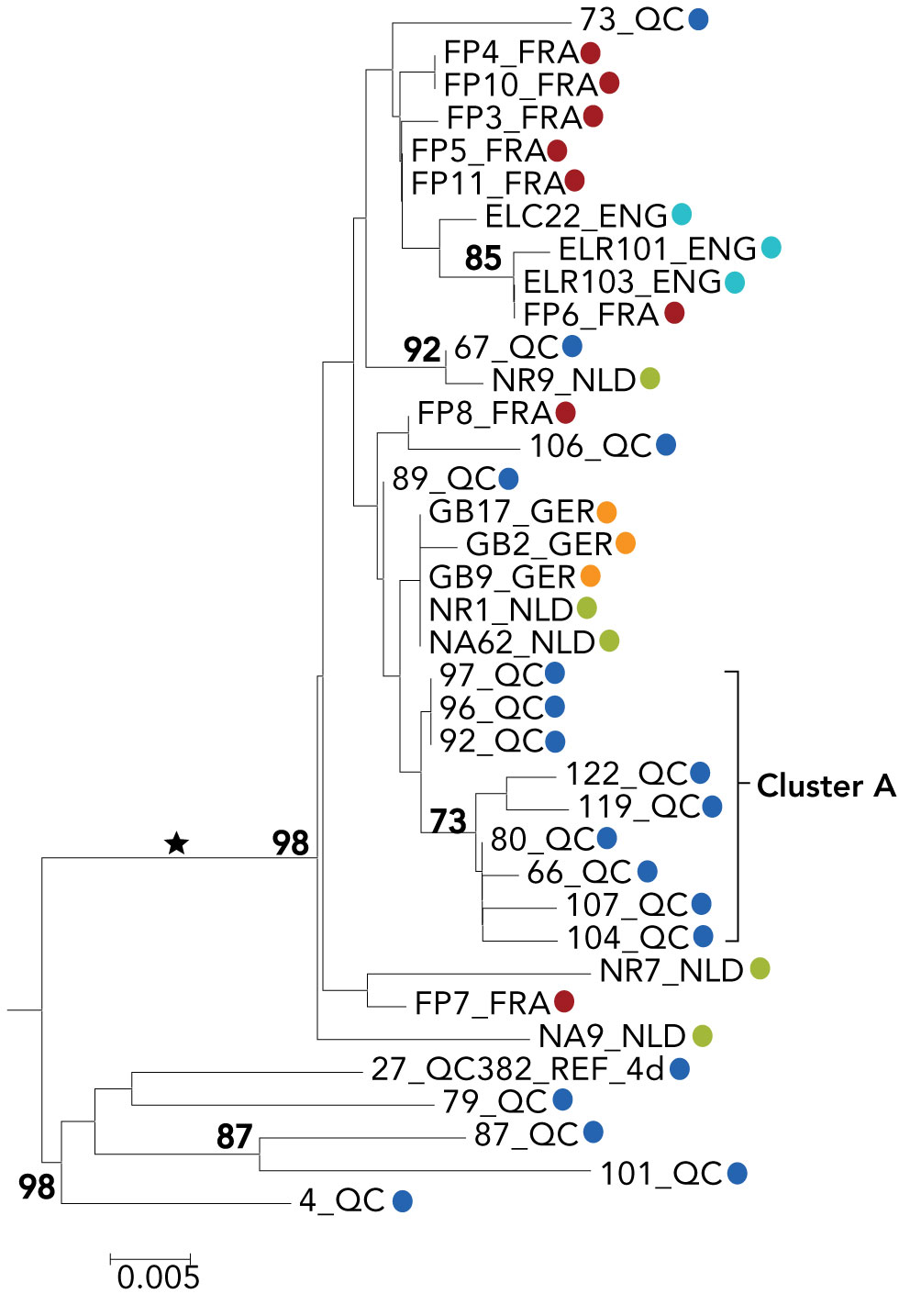
Text description: Figure A2
Figure A2: Phylogenetic tree of genotype 4d NS5B hepatitis C virus isolate sequences: cases in Quebec compared to cases in England, France, Germany and the Netherlands
The phylogenetic tree shows the genetic relatedness of different hepatitis C virus genotype 4d non-structural protein 5B sequences (nucleotide positions 8547-8982) of selected cases from Quebec, France, the Netherlands, England and Germany. The lengths of the horizontal lines or branches correspond to the genetic distance between sequences. Quebec sequences belonging to cluster A are identified. The blue dots represent Quebec, 18 cases, red dots represents France, eight cases, green dots represent the Netherlands, five cases, turquoise dots represent England, three cases, and orange dots represent England, three cases. The cross-continental monophyletic lineage is indicated by an asterisk. The numbers at nodes represent the percentage bootstrap values (only values greater than 70% are shown on nodes of non-identical sequences).
