
Ébauche de la ligne directrice - Exigences réglementaires associées à une identification numérique de drogue (DIN)
Télécharger le format de rechange
(Format PDF, 505 Ko, 29 pages)
Date de l'ébauche : 2019/01/25
| Date | Changement | Emplacement (section, paragraphe) | Nature du changement et justification |
|---|---|---|---|
| Le 25 janvier 2019 | La présente ligne directrice regroupe les politiques et les lignes directrices suivantes :
|
Changement global | Centralisation de l'information en lien avec certaines politiques et lignes directrices associées au DIN pour en faciliter l'accès |
| Le 25 janvier 2019 | Ajout de contenu : « (disponible pour usage immédiat dans les pharmacies d'hôpital et d'officine [c.-à-d. le médicament se retrouve physiquement sur les tablettes des pharmacies]), » | 6.4, paragraphe 4 |
Clarification du concept de disponibilité d'un médicament sur le marché canadien |
Avant-propos
Les lignes directrices sont destinées à guider l'industrie et les professionnels de la santé sur la façon de se conformer aux lois et aux règlements en vigueur. Les lignes directrices fournissent également aux membres du personnel des renseignements concernant la façon de mettre en œuvre le mandat et les objectifs de Santé Canada de manière juste, uniforme et efficace.
Les lignes directrices sont des outils administratifs n'ayant pas force de loi, ce qui permet une certaine souplesse d'approche. Les principes et les pratiques énoncés dans le présent document pourraient être remplacés par d'autres approches, à condition que celles-ci s'appuient sur une justification adéquate. Il faut tout d'abord discuter d'autres approches avec le programme concerné pour s'assurer qu'elles respectent les exigences des lois et des règlements applicables.
Corollairement à ce qui précède, il importe également de mentionner que Santé Canada se réserve le droit de demander des renseignements ou du matériel supplémentaire, ou de définir des conditions dont il n'est pas explicitement question dans la ligne directrice afin que le Ministère puisse être en mesure d'évaluer adéquatement l'innocuité, l'efficacité ou la qualité d'un produit thérapeutique donné. Santé Canada s'engage à justifier de telles demandes et à documenter clairement ses décisions.
Table des matières
- 1. Introduction
- 2. Objectif
- 3. Portée
- 4. Objectifs stratégiques
- 5. Définitions
- 6. Directives sur la mise en œuvre
- 6.1 Émission d'un DIN
- 6.2 État des médicaments dans la Base de données sur les produits pharmaceutiques
- 6.3 Avis de mise en marché
- 6.4 Avis de 12 mois sans vente
- 6.5 Avis de cessation de la vente
- 6.6 Annulation d'un DIN
- 6.6.1 Annulation en raison de problèmes d'innocuité
- 6.6.1.1 Annulation en raison de problèmes d'innocuité et d'efficacité selon C.01.014.6 (2) (b) du Règlement sur les aliments et drogues
- 6.6.1.2 Annulation suivant la suspension d'un avis de conformité selon C.01.014.6 (2) (c) du Règlement sur les aliments et drogues
- 6.6.1.3 Annulation à défaut de se conformer à un ordre selon C.01.014.6 (3) (a) du Règlement sur les aliments et drogues
- 6.6.1.4 Annulation suivant l'examen des résultats d'une évaluation selon C.01.014.6 (3) (b) du Règlement sur les aliments et drogues
- 6.6.2 Annulation à défaut de fournir une déclaration annuelle des médicaments selon C.01.014.6 (2) (a) du Règlement sur les aliments et drogues
- 6.6.3 Annulation : produit n'est pas ou n'est plus considéré comme étant une drogue (ou médicament) selon C.01.014.6 (1) (c) du Règlement sur les aliments et drogues
- 6.6.1 Annulation en raison de problèmes d'innocuité
- 6.7 Activités requises suivant l'annulation d'une identification numérique de drogue
- 6.8 Réémission d'un DIN par Santé Canada
- 6.9 Exportation commerciale
- 6.10 Soumission des avis à Santé Canada
Annexe A Modèle : Demande électronique de réémission d'un DIN
1. Introduction
Lorsque Santé Canada autorise la mise en marché d'un médicament au Canada, une identification numérique attribuée à une drogue (DIN) est émise au fabricant et imprimée sur les étiquettes des emballages. Un DIN indique que l'évaluation du médicament a satisfait les exigences de la Loi sur les aliments et drogues et de ses règlements et que les bénéfices liés au médicament l'emportent sur les préjudices à la santé. Les fabricants de médicaments d'ordonnance et sans ordonnance doivent obtenir un DIN avant de pouvoir les commercialiser au Canada. En plus d'une autorisation de mise en marché, l'émission d'un avis de conformité (AC) peut être nécessaire pour les nouveaux médicaments.
Le DIN attribué à un médicament est unique et peut servir d'outil facilitant les activités effectuées après la commercialisation des produits, telles que l'identification et la vérification des produits par des professionnels de la santé, les rappels de produits, les inspections et le contrôle de la qualité. Bien que l'émission d'un DIN fasse partie intégrante de l'autorisation de mise en marché d'un médicament, le DIN est la propriété de Santé Canada.
2. Objectif
La présente ligne directrice vise à :
- prêter assistance aux fabricants dans l'interprétation des exigences réglementaires associées à un DIN
- fournir aux fabricants les directives en lien avec leurs obligations de soumettre à Santé Canada les avis de changement d'état de médicaments et ce, dans les délais prescrits :
- les avis de mise en marché
- les avis de 12 mois sans vente
- les avis de cessation de la vente
3. Portée
La présente ligne directrice s'applique à tous les médicaments pour lesquels un DIN a été émis c'est-à-dire, les médicaments pour usage humain ou animal, les produits biologiques et radiopharmaceutiques et les désinfectants. La présente ligne directrice se limite aux changements affectant l'état d'un DIN. Celle-ci vise à fournir des directives en lien avec les activités suivantes :
- l'émission d'un DIN au fabricant par Santé Canada
- l'émission d'un formulaire de déclaration de médicament révisé au fabricant par Santé Canada
- la soumission par le fabricant d'avis de mise en marché auprès de Santé Canada
- la soumission par le fabricant d'avis de 12 mois sans vente auprès de Santé Canada
- la soumission par le fabricant d'avis de cessation de la vente auprès de Santé Canada
La présente ligne directrice ne vise pas à renseigner sur les activités suivantes :
- les exigences en lien avec la soumission et la gestion des présentations de drogue et des demandes d'autorisation de médicaments
- la déclaration des effets indésirables d'un médicament
- la déclaration de pénuries possibles ou réelles de médicaments sur le site Web de signalement de pénuries de médicaments géré par une tierce partie
- les frais d'utilisation et les frais à payer pour le droit de vente d'un médicament
- les licences d'établissement
- les produits qui ne détiennent pas de DIN c'est-à-dire, les dispositifs médicaux, les produits de santé naturels, les produits de santé vétérinaire, les produits antiparasitaires, les cosmétiques, les traitements expérimentaux pour les humains et les animaux et le cannabis à des fins médicales en vertu de la partie 14 du Règlement sur le cannabis
- le processus de déclaration annuelle des médicaments
4. Objectifs stratégiques
Les objectifs stratégiques qui orientent l'autorité réglementaire des activités relatives à l'émission d'un DIN et la soumission d'informations en lien avec DIN sont les suivants :
- protéger la santé et la sécurité des Canadiens contre la vente de médicaments non sécuritaires et/ou interdits
- offrir aux Canadiens de l'information exacte et fiable et ce, en temps opportun sur la disponibilité des médicaments au Canada
5. Définitions
Formulaire annuel de déclaration de médicament (FADM)
Formulaire visant à assister les fabricants à se conformer à l'article C.01.014.5 du Règlement sur les aliments et drogues, qui exige que chaque fabricant de médicament confirme, avant le 1er octobre de chaque année, que tous les renseignements fournis antérieurement concernant ce médicament sont exacts.
- Pour obtenir de plus amples renseignements sur le FDAM, consultez la Ligne directrice - Frais à payer pour le droit de vendre une drogue.
Cesser
Lorsque le fabricant cesse de façon permanente la vente d'un médicament.
Date de cessation
Lorsqu'un fabricant qui vend un médicament et qui décide d'en cesser la vente de façon permanente, la date de cessation de la vente correspond à la date de la dernière vente effectuée par le fabricant.
Lorsqu'un fabricant qui a cessé la vente d'un médicament de façon temporaire et qui, par la suite, décide d'en cesser la vente de façon permanente, la date de cessation de la vente correspond à la date à laquelle la décision de cesser la vente de façon permanente a été prise.
Drogue, au sens de l'article 2 de la Loi sur les aliments et drogues
Sont compris parmi les drogues les substances ou mélanges fabriqués, vendus ou présentés comme pouvant servir :
- au diagnostic, au traitement, à l'atténuation ou à la prévention d'une maladie, d'un désordre, d'un état physique anormal ou de leurs symptômes, chez l'être humain ou les animaux
- à la restauration, à la correction ou à la modification des fonctions organiques chez l'être humain ou les animaux
- à la désinfection des locaux où des aliments sont gardés.
Identification numérique de drogue (DIN)
Un numéro à huit chiffres généré par ordinateur qui est attribué par Santé Canada à une drogue (ou un médicament) avant sa commercialisation en application du paragraphe C.01.014.2(1) du Règlement sur les aliments et drogues.
Ce numéro identifie chaque drogue (ou médicament), en vertu du Règlement sur les aliments et drogues, vendue sous forme posologique au Canada. Il est situé sur l'étiquette de l'emballage des médicaments d'ordonnance et sans ordonnance qui ont été évalués, et dont la vente est autorisée au Canada.
Un DIN identifie de manière univoque les caractéristiques suivantes :
- le nom du produit
- le nom du fabricant
- le ou les ingrédients médicinaux
- la concentration du ou des ingrédients médicinaux
- la forme posologique
- le ou les voies d'administration
- les espèces, seulement pour les médicaments destinés aux animaux
Formulaire de déclaration de médicament (FDM)
Formulaire émis par Santé Canada en vertu de l'article C.01.014.2 (1) du Règlement sur les aliments et drogues qui contient le DIN attribué à une drogue (ou à un médicament), ainsi que quelques-uns des renseignements inclus dans la présentation de la drogue (ou du médicament).
Conformément à l'article C.01.014.3 du Règlement sur les aliments et drogues, le fabricant doit, dans les 30 jours suivant la date de la première vente de la drogue (ou du médicament), dater et signer le FDM dûment rempli et le retourner à Santé Canada, qui inclut une déclaration de l'exactitude des renseignements fournis et la date de la première vente de ce médicament.
Date limite d'une drogue (ou médicament) sous forme posologique
La date la plus antérieure parmi les suivantes :
- la date jusqu'à laquelle la drogue (ou le médicament) conserve son activité, sa pureté et ses propriétés physiques tels qu'indiqués sur l'étiquette
- la date après laquelle le fabricant recommande de ne plus utiliser la drogue (ou le médicament)
La date limite d'utilisation devrait être indiquée, au minimum, par l'année et le mois.
Étiquette
Comprend ce qui suit :
- les étiquettes apposées au contenant ou à l'emballage du médicament
- toutes notices distinctes
- les renseignements d'ordonnance
- les fiches techniques
- les renseignements sur le médicament pour le consommateur et pour le patient (c.-à-d., dépliants pour le patient)
- les carnets de suivi du patient
- les monographies de produit ou
- tout autre matériel contenant des renseignements concernant ce produit pharmaceutique en particulier
Les étiquettes des emballages créées par le fabricant peuvent être incluses dans l'emballage ou fournies au consommateur au moment de la délivrance du médicament.
Numéro de lot
Toute combinaison de lettres, de chiffres ou de lettres et de chiffres qui peuvent être utilisées pour retracer un médicament lors de sa fabrication et de sa distribution.
Fabricant
Toute personne, y compris une association ou une société de personnes, qui, sous son propre nom ou sous une marque de commerce, un dessin, une marque nominale, un nom commercial ou un autre nom, dessin ou marque soumis à son contrôle, vend un médicament. Il s'agit de la personne ou de l'entreprise à laquelle le DIN est émis. Aux fins du présent document, un fabricant peut inclure un agent autorisé à agir en son nom.
Avis de mise en marché
Avis soumis par le fabricant auprès de Santé Canada pour rapporter la date de la première vente en vertu de l'article C.01.014.3 du Règlement sur les aliments et drogues.
Drogue nouvelle, conformément à la partie C, titre 8, du Règlement sur les aliments et drogues
S'entend d'une drogue, à l'exception d'un produit de santé animale :
- qui est constituée d'une substance ou renferme une substance, sous forme d'ingrédient actif ou inerte, de véhicule, d'enrobage, d'excipient, de solvant ou de tout autre constituant, laquelle substance n'a pas été vendue comme drogue au Canada pendant assez longtemps et en quantité suffisante pour établir, au Canada, l'innocuité et l'efficacité de cette substance employée comme drogue,
- qui entre dans une association de deux drogues ou plus, avec ou sans d'autres ingrédients, qui n'a pas été vendue dans cette association particulière ou dans les proportions de ladite association pour ces drogues particulières, pendant assez longtemps et en quantité suffisante pour établir, au Canada, l'innocuité et l'efficacité de cette association ou de ces proportions employées comme drogue, ou
- pour laquelle le fabricant prescrit, recommande, propose ou déclare un usage comme drogue ou un mode d'emploi comme drogue, y compris la posologie, le mode d'administration et la durée d'action, et qui n'a pas été vendue pour cet usage ou selon ce mode d'emploi au Canada pendant assez longtemps et en quantité suffisante pour établir, au Canada, l'innocuité et l'efficacité de cet usage ou de ce mode d'emploi pour cette drogue.
Avis de conformité
Un avis émis à un fabricant suivant l'évaluation d'une présentation de drogue pour un nouveau médicament en vertu de l'article C.08.004 ou C.08.004.01 du Règlement sur les aliments et drogues.
Étiquettes portant une marque maison
Étiquette d'un médicament sans ordonnance autorisé par Santé Canada et vendu sous le nom d'un détaillant qui n'est ni le fabricant du médicament, ni la personne ou l'entreprise à laquelle le DIN a été émis.
6. Directives sur la mise en œuvre
6.1 Émission d'un DIN
Une fois qu'un médicament a été autorisé pour la vente au Canada, Santé Canada lui attribue un DIN conformément à la partie C, titre 1 du Règlement sur les aliments et drogues, permettant ainsi au fabricant de commercialiser son produit au Canada. En ce qui concerne les médicaments autorisés en vertu de la partie C, titre 8 du Règlement sur les aliments et drogues, le fabricant doit obtenir un avis de conformité (AC) et un DIN pour être autorisé à les vendre au Canada.
Avant le 13 juin 2018, seul un AC était émis pour les drogues de l'annexe C, ou produits radiopharmaceutiques, autorisées par Santé Canada. Un DIN n'était pas émis pour ces produits. Les modifications apportées au Règlement sur les aliments et drogues, entrées en vigueur le 13 juin 2018, requièrent maintenant que les fabricants de drogues de l'annexe C soumettent une demande de DIN pour obtenir un DIN pour ces produits.
- Pour obtenir de plus amples renseignements sur l'émission des DIN pour les drogues de l'annexe C, consultez la Ligne directrice : Identification numérique pour les drogues de l'annexe C (radiopharmaceutiques et trousses).
Le DIN est émis par le biais d'un formulaire de déclaration de médicament (FDM). En plus de citer le DIN, le FDM contient l'information qui est propre au médicament et ce, telle qu'autorisée par Santé Canada. Santé Canada envoie les FDM par courriel directement au fabricant.
6.1.1 Le moment de l'émission d'un DIN
6.1.1.1 Nouveaux DIN
Lorsque les fabricants demandent une autorisation pour un médicament destiné à un usage humain en vertu de la partie C, titre 8 du Règlement sur les aliments et drogues, le DIN est émis au fabricant avant l'émission de l'AC, afin qu'il puisse commencer à préparer le matériel d'étiquetage en vue de commercialiser le médicament. Cependant, le DIN n'est pas émis avant que toutes les composantes de la présentation aient été évaluées et autorisés par Santé Canada.
Lorsque les fabricants demandent une autorisation pour un médicament destiné aux animaux en vertu de la partie C, titre 8 du Règlement sur les aliments et drogues, le DIN est émis au fabricant au même moment que l'AC.
Pour les fabricants demandant une autorisation en vertu de la partie C, titre 1 du Règlement sur les aliments et drogues pour les médicaments pour usage humain ou animal, un AC n'est pas émis. Le DIN, émis par le biais d'un FDM, représente l'autorisation pour la vente du médicament au Canada.
6.1.1.2 Formulaire de déclaration de médicament révisé
Le changement d'une ou plusieurs caractéristiques d'un médicament, tel qu'indiqué dans la définition d'un DIN à la section 5, doit être autorisé avant que le FDM puisse être révisé. Les changements proposés doivent être soumis à Santé Canada par le biais d'une présentation de drogue ou d'une demande d'autorisation.
Pour tout changement à un médicament autorisé pour un usage humain ou animal en vertu de la partie C, titre 8, la révision du FDM, portant la même séquence de chiffres que le DIN ayant été émis au fabricant auparavant, est effectuée après que l'AC est émis. Si le médicament est autorisé, Santé Canada émettra soit un nouveau DIN ou un FDM révisé portant la même séquence de chiffres que le DIN ayant été émis au fabricant.
Pour tout changement à un médicament autorisé pour un usage humain ou animal en vertu de la partie C, titre 1, un FDM révisé portant la même séquence de chiffres que le DIN ayant été émis au fabricant, peut être émis. Le FDM représente l'autorisation de mise en marché; un AC n'est émis pas pour ces médicaments.
Le tableau 1 ci-dessous indique les changements pouvant être apportés aux caractéristiques d'un médicament qui nécessiteraient soit l'émission d'un nouveau DIN ou l'émission d'un FDM révisé portant la même séquence de chiffres que le DIN ayant été émis au fabricant.
| Changement dans les caractéristiques | Nouveau DIN | FDM révisé portant la même séquence de chiffres que celle du DIN ayant été émis au fabricant |
|---|---|---|
| Nom du produit | - | ✓ |
| Nom du fabricant | - | ✓ |
| Ingrédients médicinal | ✓ | - |
| Concentration de l'ingrédient médicinal | ✓ | - |
| Forme posologique | ✓ | - |
| Voie d'administration | ✓ | ✓Tableau 1 Note de bas de page * |
| Espèce, seulement pour les médicaments destinés aux animaux | - | ✓ |
| ||
6.1.2 Attribution d'un DIN en fonction du nom du produit
Le nom de produit est proposé par un fabricant afin qu'il puisse en faire la commercialisation et la publicité, lorsqu'applicable. Avant leur autorisation, Santé Canada évalue le nom de chaque produit inclus dans une présentation de drogue ou une demande d'autorisation pour un médicament.
- Pour obtenir de l'information sur l'évaluation des noms de produits de médicaments pour usage humain, consultez la :
Si un fabricant désire commercialiser un médicament autorisé, c'est-à-dire pour lequel un DIN a déjà été attribué, et ce, sous des noms différents, un nouveau DIN sera attribué pour chaque nouveau nom.
- Pour plus d'information sur la soumission d'une présentation de drogue pour l'ajout d'un nouveau nom de produit, consultez la Foire aux questions - Ligne directrice à l'intention de l'industrie - Examen des marques nominatives de médicament
- Pour toutes questions concernant les noms de produit pour les médicaments destinés aux animaux, communiquez avec la Direction des médicaments vétérinaires à hc.vdd.skmd.so-dgps.dmv.cp.sc@canada.ca
Un nouveau DIN ne sera pas émis à un fabricant désirant commercialiser un médicament sans ordonnance, préalablement autorisé par Santé Canada, et portant une étiquette de marque maison, telle que définie dans la section 5, si le nom de produit demeure inchangé.
Le tableau 2 ci-dessous fournit un exemple de deux produits avec le même DIN.
| DIN | ZZZZZZZZ | ZZZZZZZZ |
|---|---|---|
| Nom du produit | Oméprazole | Oméprazole |
| Nom du fabricant | Compagnie pharmaceutique | Compagnie pharmaceutique |
| Détaillant | Pharmacie1 | Pharmacie2 |
L'ajout ou la modification d'éléments spécifiques à une étiquette portant une marque maison (p. ex. éléments graphiques, couleurs, police, etc.), préalablement autorisée par Santé Canada, doivent être évalués et autorisés avant d'être introduite sur le marché. Ceci signifie que les notifications à la Division des politiques sur les présentations et renseignements (DPPR) ne seront plus acceptées pour le prolongement d'une marque maison.
- Pour plus d'information sur la soumission d'étiquettes supplémentaires pour un médicament vendu sans ordonnance et autorisé pour un usage humain, consultez la section 5.9 du Document d'orientation : Questions et réponses : Le règlement sur l'étiquetage en langage clair pour les médicaments vendus sans ordonnance
6.1.3 Émission de plusieurs DIN pour adresser les problèmes d'innocuité
Dans le but de prévenir des erreurs de posologie potentiellement graves, Santé Canada peut, dans certaines circonstances, émettre plusieurs DINs pour un médicament dont différents formats sont autorisés pour une même concentration, tels que des formats à dose unique. Ceci peut également s'appliquer aux stylos et aux auto-injecteurs préremplis de même concentration et disponibles en formats variés.
Lorsqu'une présentation de drogue ou une demande d'autorisation de médicament est soumise et autorisée pour une seringue préremplie à dose unitaire dont la concentration est la même mais, dont les formats diffèrent, un DIN sera émis pour chacun des formats. Par exemple, 2 DIN seront émis pour une seringue préremplie de 10 mg/ml offerte en dose unitaire de 1 ml et de 2 ml. Pour les produits qui sont présentement disponibles sur le marché, Santé Canada adressera ces cas de façon individuelle.
Le développement de cette politique se poursuivra lorsque Santé Canada aura acquis davantage d'expérience quant à son application.
6.2 État des médicaments dans la Base de données sur les produits pharmaceutiques
Une fois que le DIN ou que le DIN et l'AC ont été émis au fabricant, Santé Canada publie les renseignements associés à ce médicament, y compris le DIN et son état, dans la Base de données sur les produits pharmaceutiques (BDPP). L'état d'un médicament dans la BDPP indique sa disponibilité sur le marché canadien. En vertu du Règlement sur les aliments et drogues, le fabricant est tenu d'informer Santé Canada de tout changement concernant l'état d'un médicament.
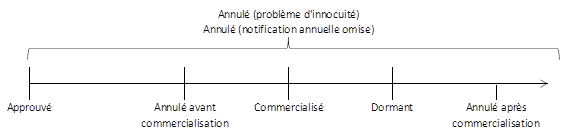
Figure 1 : Ordre chronologique des états pouvant possiblement être associés à un médicament et apparaissant dans la BDPP
La figure 1 illustre l'ordre chronologique de cinq états pouvant possiblement être associés à un médicament. De gauche à droite : approuvé, annulé avant commercialisation, commercialisé, dormant et annulé après commercialisation. Les états annulé (problème d'innocuité) et annulé (déclaration annuelle omise) apparaissent au haut de la ligne chronologique pour illustrer que l'un ou l'autre de ces événements peut se produire en tout temps suivant l'émission d'un DIN.
Chacun des états est décrit ci-après :
- Approuvé : fait référence à un DIN qui a été autorisé pour la vente au Canada, mais qui n'est pas encore commercialisé au Canada. Le DIN est considéré comme étant actif.
- Annulé (avant commercialisation) : fait référence à un DIN qui a été annulé avant sa commercialisation au Canada. Le DIN est considéré comme étant inactif.
- Commercialisé : fait référence à un DIN présentement vendu au Canada. Le DIN est considéré comme étant actif.
- Dormant : fait référence à un DIN commercialisé au Canada, qui n'a été vendu pendant au moins 12 mois consécutifs. Le DIN est considéré comme étant actif, puisque le médicament demeure autorisé pour la vente au Canada et peut être commercialisée à nouveau.
- Annulé après commercialisation : fait référence à un DIN qui a été annulé suivant la cessation de la vente d'un médicament part le fabricant. Le DIN est considéré comme étant inactif.
- Annulé (problème d'innocuité) : fait référence à un DIN annulé en vertu des alinéas du Règlement sur les aliments et drogues mentionnés ci-dessous. Dans tous ces cas, le DIN est considéré comme étant inactif.
- en vertu de l'alinéa C.01.014.6 (2) (b) du Règlement sur les aliments et drogues et à défaut d'avoir fourni les données probantes relatives à l'innocuité et à l'efficacité du médicament, conformément à l'article C.01.013 du Règlement sur les aliments et drogues
- en vertu de l'alinéa C.01.014.6 (2) (b) du Règlement sur les aliments et drogues et suivant de la suspension d'un Avis de conformité, conformément à l'article C.08.006
- en vertu de l'alinéa C.01.014.6 (3) (a) du Règlement sur les aliments et drogues et à défaut de s'être conformé à l'ordre émis conformément à l'article 21.31 de la Loi sur les aliments et drogues, demandant d'effectuer une évaluation et d'en fournir les résultats
- en vertu de l'alinéa C.01.014.6 (3) (b) du Règlement sur les aliments et drogues et suivant l'évaluation des résultats d'une évaluation, fournis en réponse à l'ordre émis conformément à l'article 21.31 de la Loi sur les aliments et drogues.
- Annulé (déclaration annuelle omise) : fait référence à un DIN qui a été annulé, à défaut d'avoir soumis le Formulaire de déclaration annuelle des médicaments, conformément à l'alinéa C.01.014.6 (2) (a) du Règlement sur les aliments et drogues. Dans ce cas-ci, le DIN est considéré comme étant inactif.
6.3 Avis de mise en marché
Conformément à l'article C.01.014.3 du Règlement sur les aliments et drogues, le fabricant se doit d'informer Santé Canada lorsqu'un médicament, pour lequel un nouveau DIN a été émis, a été vendu pour la première fois au Canada. Un fabricant doit soumettre un FDM dûment rempli à Santé Canada dans les 30 jours suivant la première vente du médicament. Le FDM doit être dûment rempli, signé et daté. Toutes les pages du FDM doivent être soumises à Santé Canada.
Si le FDM a été révisé par Santé Canada (voir la section 6.1.1.2 pour plus d'information), le fabricant se doit d'informer Santé Canada lorsque le médicament est a été vendu pour la première fois sous le changement autorisé. Un fabricant doit présenter un FDM dûment rempli à Santé Canada dans les 30 jours suivant la première vente du médicament ayant fait l'objet du changement. Le FDM doit être dûment rempli, signé et daté. Toutes les pages du FDM doivent être soumises à Santé Canada.
6.3.1 Complétion de l'avis de mise en marché
Un avis de mise en marché inclus les documents ci-dessous :
- une lettre d'accompagnement
- un FDM dûment rempli et signé
- le matériel d'étiquetage, lorsqu'applicable (voir le tableau 3)
Ce n'est que lorsqu'un avis de mise en marché complet et dûment rempli est reçu et traité que l'état du médicament dans la BDPP peut être modifié à Commercialisé.
Les sections ci-dessous visent à informer le fabricant sur :
- la façon de remplir correctement le FDM
- les circonstances dans lesquelles le matériel d'étiquetage doit être soumis
6.3.1.1 Partie I du Formulaire de déclaration de médicament
La partie I du FDM présente les renseignements inclus dans la BDPP. Il incombe au fabricant de vérifier les renseignements sur le FDM au moment de sa réception. En cas d'erreurs dans les coordonnées de l'entreprise à qui le DIN a été émis ou fabricant, de l'agent ou du ou des importateurs soient l'adresse postale, le nom de la personne à contacter, le numéro de téléphone, le numéro de télécopieur et l'adresse électronique, le fabricant doit biffer les renseignements erronés et inscrire les renseignements exacts dans les espaces prévus à cette fin.
Aucune modification ne peut être apportée au nom de l'entreprise ou du fabricant à qui le DIN a été émis, le nom du produit, la forme posologique, la voie d'administration, les ingrédients médicinaux, la concentration et les espèces pour les médicaments destinés aux animaux inscrits sur le FDM. Le fabricant doit soumettre une présentation de drogue ou une demande d'autorisation du médicament pour que ces changements soient effectués.
6.3.1.2 Partie II du Formulaire de déclaration de médicament
La partie II du FDM contient les renseignements fournis par le fabricant dans le cadre de l'avis de mise en marché.
Les renseignements ci-après doivent être fournis dans la partie II.
- La date à laquelle le médicament a été vendu pour la première fois au Canada suivant :
- son autorisation par Santé Canada ou
- l'autorisation d'un changement pour lequel un FDM révisé a été émis ou
- une période d'au moins 12 mois consécutifs pendant laquelle il n'y a eu aucune vente et pour laquelle un avis de 12 mois sans vente a été soumis à Santé Canada
- Le représentant autorisé (titre, signature) :
- Toute personne désignée par le fabricant pour agir en son nom
- La date :
- La date à laquelle le FDM a été rempli et signé
6.3.1.3 Soumission des étiquettes avec un avis de mise en marché
Dans certaines circonstances, le matériel d'étiquetage doit être soumis avec l'avis de mise en marché.
En 2014, la publication du Règlement modifiant le Règlement sur les aliments et drogues (étiquetage, emballage et marques nominatives des drogues pour usage humain) (Règlement sur l'étiquetage en langage clair) a abrogé l'exigence de soumettre les étiquettes commercialisées une fois que le médicament a été mis en marché, tel qu'indiqué dans le Règlement sur les aliments et drogues.
Par conséquent, pour déterminer si une copie des étiquettes commercialisées doit être soumise avec un avis de mise en marché pour un médicament, conformément au Règlement sur l'étiquetage en langage clair, consultez le tableau 3.
| Date à laquelle l'activité de réglementation (p.ex. PNM, DDIN) a été soumise et autorisée par Santé Canada | |||
|---|---|---|---|
| Médicament | Avant le 13 juin 2015 | Entre le 13 juin 2015 et le 13 juin 2017 | Après le 13 juin 2017 |
| D'ordonnance | Oui | Non | Non |
| Administré ou délivré par un professionnel de la santé (éthique) | Oui | Non | Non |
| Sans ordonnance | Oui | Oui | Non |
| Désinfectant | Oui | Oui | Oui |
| D'ordonnance et sans ordonnance destinés aux animaux | Oui | Non | Non |
6.4 Avis de 12 mois sans vente
Il est possible que le fabricant n'est pas vendu un médicament, à certains moments, lors de sa commercialisation. Si l'une ou l'autre de ces périodes dure au moins 12 mois consécutifs, le fabricant se doit, en vertu de l'article C.01.014.71 et du sous-alinéa C.01.014.5(1)(a)(ii) du Règlement sur les aliments et drogues, d'en informer Santé Canada. Les instances où le fabricant se doit d'informer Santé Canada de ces périodes sont décrites dans les sections 6.4.1 et 6.4.2.
Après avoir reçu un avis complet et traité l'information, Santé Canada modifie l'état dans la BDPP pour indiquer de ce médicament est Dormant. Un produit considéré comment étant dormant demeure autorisé par Santé Canada et peut être commercialisé à nouveau.
Lorsque 12 mois se sont écoulés depuis la dernière vente du médicament au Canada, le fabricant se doit d'informer Santé Canada. Les trois conditions suivantes doivent être remplies :
- un AC ou un DIN et un AC ont été émis pour ce médicament
- le médicament a été commercialisée au Canada et
- le médicament n'a pas été vendu sur le marché canadien pendant une période de 12 mois consécutifs
Dans le cas d'un médicament qui n'a connu aucune vente en raison d'une faible demande causée, par exemple, par une faible population de patients visée par ce traitement, le fabricant se doit d'informer Santé Canada que le médicament est considéré comme étant dormant, conformément à l'article C.01.014.71 et au sous-alinéa C.01.014.5(1)(a)(ii) du Règlement sur les aliments et drogues, s'il maintient un inventaire de ce médicament et si le médicament est disponible sur le marché canadien. Un médicament qui est disponible sur le marché canadien signifie qui peut être dispensé immédiatement par les pharmacies d'hôpital et d'officine. En d'autres mots, le médicament se trouve sur les tablettes des pharmacies. Le fabricant peut donner des détails sur la situation au moment d'en informer Santé Canada.
Cependant, dans une telle situation, Santé Canada peut choisir de conserver l'état du médicament dans la BDPP comme étant Commercialisé afin d'éviter des répercussions sur les plans de traitement des patients. Si le médicament est vendu sur le marché, l'obligation d'en informer Santé Canada, conformément à l'article C.01.014.71 ou au sous-alinéa C.01.014.5(1)(a)(ii) du Règlement sur les aliments et drogues, ne s'applique plus.
6.4.1 Avis de douze mois sans vente dans les 30 jours civils
Les fabricants sont encouragés à informer Santé Canada lorsqu'un médicament n'a pas été vendu pendant au moins 12 mois consécutifs et ce, dans les 30 jours suivant cette période. Cela permet à Santé Canada ainsi qu'aux patients, aux professionnels de la santé et à d'autres intervenants du système de santé d'avoir des renseignements clairs et à jour sur les médicaments disponibles sur le marché canadien.
Toutefois, en vertu de l'article C.01.014.71 du Règlement sur les aliments et drogues, les fabricants de médicaments pour usage humain, listés ici-bas, qui n'ont pas vendus pour une période de 12 mois consécutifs sont dans l'obligation d'en d'informer Santé Canada dans les 30 jours civils. Ces médicaments sont :
- les médicaments inclus aux annexes I, II, III, IV ou V de la Loi réglementant certaines drogues et autres substances
- les médicaments d'ordonnance
- les produits inclus aux annexes D et C de la Loi
- les médicaments pouvant être vendus sans ordonnance, mais qui sont administrés uniquement sous la supervision d'un praticien (p. ex. les solutions d'hémodialyse, l'épinéphrine pour les réactions allergiques graves, les produits de contraste pour imagerie par résonance magnétique (IRM), l'insuline et les vaccins), également connus sous le nom de médicaments éthiques
Le fabricant doit envoyer l'avis de 12 mois sans vente par l'entremise d'une lettre officielle, portant l'entête de l'entreprise, dûment signée par un agent autorisé. L'avis doit être envoyé par voie électronique, tel qu'il est indiqué dans la section 6.9.
Ce n'est que lorsqu'un avis de 12 mois sans vente complet et dûment rempli est reçu et traité que l'état du médicament dans la BDPP peut être modifié à Dormant.
6.4.2 Soumission de l'avis de 12 mois sans vente pour tous les médicaments par le biais du Formulaire de déclaration annuelle de médicament
Les fabricants de médicaments ayant obtenu un DIN, en vertu du sous-alinéa C.01.014.2(1) du Règlement sur les aliments et drogues, qui sont commercialisés, doivent informer Santé Canada si l'état de ces médicament doit être changé à Dormant au moment de la soumission du Formulaire de déclaration annuelle des médicaments (FDAM).
Les directives pour compléter et soumettre le FDAM sont incluses dans la trousse de la Déclaration annuelle des médicaments envoyée en juin de chaque année par Santé Canada.
Ce n'est que lorsqu'un FDAM complet et dûment rempli est reçu et traité que l'état peut être modifié dans la BDPP pour indiquer que le médicament est Dormant. La date associée à l'état de dormance sera la date apposée sur le FDAM signé.
6.4.3 Reprise de la vente après l'état de dormance
Si le fabricant reprend la vente d'un médicament dont l'état était Dormant, celui-ci doit soumettre, dans les 30 jours suivant la reprise de la vente de ce médicament, un FDM signé et daté. La date de reprise de la vente doit être indiquée sur le FDM. Le fabricant ne doit pas utiliser la date à laquelle le médicament a été commercialisé pour la première fois. La soumission des étiquettes commercialisées n'est pas requise lorsqu'un médicament est vendu à nouveau après un état de dormance.
6.5 Avis de cessation de la vente
Le fabricant doit soumettre un avis de cessation de vente dans les 30 jours suivant la cessation de la vente de la drogue, conformément à l'article C.01.014.7 du Règlement sur les aliments et drogues. Santé Canada annule le DIN après la réception et le traitement d'un avis complet, conformément à l'alinéa C.01.014.6 (1) (a) du Règlement sur les aliments et drogues. Le fabricant doit se conformer aux obligations associées à ce médicament avant l'annulation du DIN et ce, jusqu'à l'échéance du dernier lot distribué ou de la plus longue période précisée dans le Règlement sur les aliments et drogues. Veuillez consultez la section 6.7 pour plus d'information sur les activités requises suivant l'annulation d'un DIN.
Ce n'est que lorsqu'un avis de cessation de la vente complet et dûment rempli est reçu et traité que l'état est modifié dans la BDPP pour indiquer que le médicament a été Annulé. La date associée à la cessation de la vente du médicament est la date indiquée par le fabricant dans la lettre. Si la date de cessation n'est pas incluse dans la lettre, la date de la lettre sera plutôt utilisée.
- Si le médicament n'a jamais été commercialisé, c'est-à-dire que son état est Approuvé dans la BDPP, l'état sera modifié pour indiquer que le médicament est Annulé après commercialisation.
- Si le médicament est commercialisé ou a déjà été commercialisée, c'est-à-dire que son état est Commercialisé ou Dormant dans la BDPP, l'état sera modifié pour indiquer que le médicament est Annulé après commercialisation. La date limite d'utilisation du dernier lot distribué au Canada, le numéro de lot et la date d'annulation du DIN doivent être fournis par le fabricant pour être affichés sur la BDPP.
Santé Canada enverra une confirmation au fabricant ou à l'agent désigné indiquant que l'annulation du DIN a été traitée.
Seuls les médicaments, dont le DIN est considéré actif, peuvent faire renvoi à d'autres médicaments déjà autorisés. De plus, les titulaires de licence doivent consulter la section 2.5.4.1 de la Ligne directrice : Traitement administratif des présentations et des demandes concernant les médicaments destinés aux humains ou les désinfectants suivant l'annulation d'un DIN par leur concédant afin de se familiariser avec les répercussions sur leur médicaments qui font l'objet d'un contrat de licence, traité de façon administrative par Santé Canada.
6.5.1 Cessation de la vente après l'état de dormance
Après avoir informé Santé Canada de la période de 12 mois sans vente, un fabricant peut décider de cesser la vente de ce médicament. Dans les cas où l'état du médicament est considéré comme étant Dormant, la date de cessation doit être ultérieure à la date où le médicament a été déclaré comme étant Dormant.
6.6 Annulation d'un DIN
Plusieurs raisons peuvent mener à annulation d'un DIN par Santé Canada. Ces raisons sont décrites dans l'article C.01.014.6 du Règlement sur les aliments et drogues.
6.6.1 Annulation en raison de problèmes d'innocuité
6.6.1.1 Annulation en raison de problèmes d'innocuité et d'efficacité selon C.01.014.6 (2) (b) du Règlement sur les aliments et drogues
Santé Canada peut annuler DIN lorsqu'un fabricant ne fournit pas suffisamment de données probantes concernant l'innocuité et l'efficacité liées à une utilisation recommandée pour un médicament.
Lorsqu'il est déterminé qu'une annulation de DIN est justifiée, Santé Canada modifiera l'état dans la BDPP pour indiquer que le médicament est Annulé (problème d'innocuité).
6.6.1.2 Annulation suivant la suspension d'un avis de conformité selon C.01.014.6 (2) (c) du Règlement sur les aliments et drogues
Tel que décrit à l'alinéa C.08.002 (1) (c) du Règlement sur les aliments et drogues, un médicament ne peut plus être vendu si l'AC est suspendu par Santé Canada. L'interdiction de vendre entre en vigueur dès que l'AC a été suspendu et s'applique aux fabricants ainsi qu'à tout autre parti tels que les grossistes, les détaillants, les pharmaciens et les praticiens.
Suivant la suspension d'un AC, Santé Canada peut annuler le DIN conformément à l'alinéa C.01.014.6 (2) (c) du Règlement sur les aliments et drogues.
Suivant la décision d'annuler le DIN, Santé Canada modifiera l'état dans la BDPP pour indiquer que le médicament a été Annulé (problème d'innocuité). La base de données des AC sera également mise à jour pour indiquer que l'AC a été suspendu.
6.6.1.3 Annulation à défaut de se conformer à un ordre selon C.01.014.6 (3) (a) du Règlement sur les aliments et drogues
Pour permettre à Santé Canada de réglementer un médicament de façon efficace et effective, Santé Canada a le pouvoir d'ordonner au fabricant d'effectuer des évaluations, de compiler des renseignements, de faire des essais ou des études ou de surveiller l'expérience acquis sur le médicament et de lui fournir les résultats en vertu de l'article 21.31 de la Loi sur les aliments et drogues.
- Pour plus d'information sur le pouvoir de Santé Canada d'exiger une évaluation en vertu de l'article 21.31 de la Loi sur les aliments et drogues ainsi que sur le pouvoir de demander un essai, des études, etc., consultez le document Modifications à la Loi sur les aliments et drogues : Guide pour l'application des nouveaux pouvoirs : Pouvoir d'exiger et de communiquer des renseignements, Pouvoir d'exiger la modification d'une étiquette, Pouvoir d'ordonner un rappel.
Si, en vertu de l'article 21.31 de la Loi sur les aliments et drogues, le fabricant ne se conforme pas à l'ordre de fournir l'information demandée, Santé Canada peut annuler le DIN conformément à l'alinéa C.01.014.6 (3)(a) du Règlement sur les aliments et drogues.
Suivant de la décision d'annuler le DIN, Santé Canada modifiera l'état dans la BDPP pour indiquer que le médicament est Annulé (problème d'innocuité).
6.6.1.4 Annulation suivant l'examen des résultats d'une évaluation selon C.01.014.6 (3) (b) du Règlement sur les aliments et drogues
Si, suivant l'évaluation de renseignements fournis en réponse à un ordre, en vertu de l'article 21.31 de la Loi sur les aliments et drogues, il a été déterminé que les risques de préjudices à la santé l'emportent sur les bénéfices du médicament, Santé Canada peut annuler le DIN conformément à l'alinéa C.01.014.6 (3)(b) du Règlement sur les aliments et drogues.
Suivant la décision d'annuler le DIN, Santé Canada modifiera l'état dans la BDPP pour indiquer que le médicament a été Annulé (problème d'innocuité).
6.6.2 Annulation à défaut de fournir une déclaration annuelle des médicaments selon C.01.014.6 (2) (a) du Règlement sur les aliments et drogues
Chaque année, les fabricants doivent fournir à Santé Canada une copie signée de leur Formulaire de déclaration annuelle des médicaments (FDAM). Le FDAM permet aux fabricants d'attester que tous les renseignements concernant leurs médicaments sont exacts et de fournir des mises à jour à Santé Canada.
Avant le 1er octobre de chaque année, Santé Canada communique avec tous les fabricants qui n'ont pas retourné une copie signée du FDAM pour leur rappeler de se conformer à leurs obligations réglementaires.
Si Santé Canada n'a pas reçu le FDAM au plus tard le 1er octobre, conformément à l'article C.01.014.5 du Règlement sur les aliments et drogues, Santé Canada peut annuler les DIN. Un dernier avis écrit sera émis au fabricant pour l'informer que ses DIN seront annulés, conformément à l'alinéa C.01.014.6 (2) (a) du Règlement sur les aliments et drogues, et qu'il ne peut plus vendre ces médicaments, conformément à l'alinéa C.01.014 (1) du Règlement sur les aliments et drogues.
Par la suite, Santé Canada modifiera l'état dans la BDPP pour indiquer que le médicament a été Annulé (déclaration annuelle omise).
6.6.3 Annulation : produit n'est pas ou n'est plus considéré comme étant une drogue (ou médicament) selon C.01.014.6 (1) (c) du Règlement sur les aliments et drogues
Santé Canada peut annuler un DIN lorsqu'il a déterminé que le produit n'est pas ou n'est plus considéré comme étant une drogue (ou un médicament) en vertu du Règlement sur les aliments et drogues.
Dans une telle situation, Santé Canada expliquera par écrit au fabricant que le produit est reclassé et qu'il n'est plus réglementé en tant que drogue (ou médicament) en vertu du Règlement sur les aliments et drogues.
Santé Canada fournira au fabricant la date à laquelle l'état du médicament sera modifié et la date à laquelle le DIN sera annulé. Le cas échéant, des renseignements sur le ou les règlements auxquels le produit sera assujetti seront fournis au fabricant pour lui pour permettre de commercialiser le produit sur le marché canadien.
Après le changement de classification du produit, Santé Canada annulera le DIN et celui-ci sera retirer de la BDPP.
6.7 Activités requises suivant l'annulation d'une identification numérique de drogu
Lorsqu'un DIN est annulé en vertu de l'article C.01.014.6 du Règlement sur les aliments et drogues, aucune autre vente ne peut être effectuée par le fabricant puisque l'alinéa C.01.014 (1) du Règlement sur les aliments et drogues interdit à un fabricant de commercialiser une drogue qui n'a pas de DIN.
Afin de ne pas imposer un fardeau excessif à l'industrie, Santé Canada peut permettre aux grossistes, aux détaillants, aux pharmaciens et aux praticiens de continuer à vendre ou à distribuer les inventaires restant du médicament après l'annulation de son DIN, si :
- la date limite d'utilisation du ou des lot n'est pas dépassée et
- l'annulation du DIN n'est pas reliée à un problème de santé ou d'innocuité
Si le DIN est annulé, le médicament ne peut plus être importé au Canada à des fins commerciales.
Si Santé Canada est informé de tout risque ou de toute activité non conforme liée à un médicament dont le DIN a été annulé, les mesures appropriées seront mises en place pour atténuer le risque, conformément à la Politique de conformité et d'application (POL-0001).
Les scénarios ci-dessous illustrent quelques-unes des activités qu'un fabricant doit poursuivre suivant l'annulation d'un DIN. Les fabricants doivent consulter les règlements pour obtenir tout détail sur leurs obligations.
Scénario no 1 : Mises à jour des renseignements sur l'innocuité du médicament dans la monographie de produit
Même si le DIN a été annulé, le fabricant doit continuer de soumettre les présentations de drogue ou les demandes d'autorisation appropriées pour ajouter ou changer toute information relative à l'innocuité, précisément l'information reliée aux contre-indications, aux mises en garde et aux précautions ou aux effets indésirables, nécessaires lorsque de nouveaux renseignements sont disponibles à cet effet et ce, jusqu'à ce que tous les lots du médicament sur le marché soient expirés.
Scénario no 2 : Déclaration des effets indésirables
Les exigences voulant que les fabricants de même que les grossistes et les distributeurs tiennent à jour leurs dossiers d'effets indésirables respectivement en vertu des articles C.01.020 et C.02.022 du Règlement sur les aliments et drogues continuent de s'appliquer après l'annulation du DIN. Bien que le fabricant ne soit pas tenu de déclarer les nouveaux cas d'effets indésirables reçus après la cessation de la vente d'un médicament, Santé Canada encourage fortement la déclaration de tous effets indésirables graves et peut demander au fabricant de fournir ces renseignements. D'autres renseignements sur les exigences en matière de déclaration pour les médicaments à usage humain sont disponibles dans le document : Déclaration des effets indésirables des produits de santé commercialisés - Document d'orientation à l'intention de l'industrie.
Scénario no 3: Vente, tenue de dossiers et déclaration
Un fabricant cesse la vente d'un médicament le 28 février et informe Santé Canada le même jour. Santé Canada annule le DIN. Toutefois, le médicament est disponible sur le marché et la date limite d'utilisation du dernier lot est le 30 décembre.
Les grossistes, les détaillants, les pharmaciens et les praticiens peuvent continuer à vendre ou à distribuer le médicament jusqu'au 30 décembre tout en sachant que des lots étaient en leur possession avant l'annulation du DIN. Toutefois, un importateur ne peut continuer à importer à des fins commerciales un médicament dont le DIN a été annulé.
Les grossistes, les distributeurs et les importateurs doivent se conformer à toutes les exigences associées à la tenue de dossiers en vertu du Règlement sur les aliments et drogues, y compris l'article C.02.022 du Règlement sur les aliments et drogues, et ce, pour tous les lots de médicament existant sur le marché, incluant ceux introduit sur le marché après l'annulation du DIN, soit entre le 28 février et le 30 décembre. En vertu de l'article C.02.022 du Règlement sur les aliments et drogues, les dossiers doivent être conservés pendant un 1 an après la date limite d'utilisation du dernier lot, à moins que la licence d'établissement ne précise une autre période.
Scénario no 4 : Mise à jour de l'étiquetage
Dans le cas où un fabricant annule un DIN associé à une concentration d'un médicament (p. ex. médicament X, 10 mg) tout en conservant les DIN associés aux autres concentrations de ce même médicament (p. ex. médicament X, 20 mg, 40 mg, 80 mg), le fabricant doit soumettre une présentation de drogue ou un demande d'autorisation pour retirer de l'étiquetage les renseignements ayant trait au DIN annulé (c'est-à-dire, la monographie du produit, l'encart, les renseignements d'ordonnance, etc.) et ce, après la date limite d'utilisation de tous les lots du médicament disponibles sur le marché.
6.8 Réémission d'un DIN par Santé Canada
Dans certaines circonstances, Santé Canada peut réémettre la même séquence de chiffres que celle du DIN originellement émis pour le même médicament, après que celui-ci ait été annulé.
Le fabricant doit communiquer avec le Bureau des présentations et de la propriété intellectuelle (BPPI) pour connaître les exigences applicables à une présentation de drogue ou une demande d'autorisation d'un médicament avant de procéder à la remise en marché de ce médicament.
La demande doit être envoyée par courriel au BPPI à HC.DIN.SC@canada.ca en utilisant le modèle à l'annexe A.
Les renseignements obtenus par le fabricant seront envoyés au bureau d'évaluation approprié pour déterminer quel type de présentation de drogue ou de demande d'autorisation doit être soumise (p. ex. (S)P(A)DN, DDIN(PB)(PD), CPA) pour réémettre la même séquence de chiffres que celle du DIN originellement émis pour le même médicament, le cas échéant. Les frais associés à la présentation de drogue ou à la demande d'autorisation de même que les délais d'évaluation vont s'appliquer.
À la discrétion de Santé Canada, les DIN peuvent être réémis sans soumission d'une présentation de drogue ou de demande d'autorisation ni frais associés, si le fabricant atteste qu'aucune modification n'a été apportée au médicament ou à son étiquetage, depuis l'annulation du DIN.
Le médicament pour lequel le fabricant souhaite obtenir la réémission du DIN est assujetti à toutes les exigences réglementaires existantes, incluant une évaluation de nom de produit à présentation et à consonance semblables. Si un problème potentiel lié aux présentations et aux consonances semblables est identifié, il est possible que le nom du produit soit modifié. Consultez la Ligne directrice à intention de l'industrie - Examen des marques nominatives de médicament pour obtenir de plus amples renseignements sur les évaluations de nom de produits.
Si les renseignements fournis par le fabricant sont acceptables, le FDM portant la même séquence de chiffres que celle du DIN originellement sera émis au fabricant. L'état du DIN dans la BDPP sera modifié pour indiquer que le médicament est Approuvé. Le fabricant se devra d'informer Santé Canada lorsque le produit sera commercialisé, conformément à l'article C.01.014.3 du Règlement sur les aliments et drogues. Consultez la section 6.3 ci-dessus pour obtenir des renseignements supplémentaires sur les avis de mise en marché.
6.9 Exportation commerciale
Lorsqu'un fabricant cesse la vente d'un médicament destiné à la consommation au Canada, mais continue de le commercialiser et de l'exporter, l'exportation avec ou sans l'invocation de l'article 37 de la Loi sur les aliments et drogues déterminera si la vente du médicament est considérée comme ayant cessé au Canada.
- Pour obtenir un complément d'information sur l'article 37 de la Loi sur les aliments et drogues, consultez le document : L'intention d'invoquer l'article 37 de la Loi des aliments et drogues pour les produits destinés à l'exportation.
6.9.1 Exportation sans l'invocation de l'article 37
Pour exporter un médicament sans l'invocation de l'article 37 de la Loi sur les aliments et drogues, le fabricant doit détenir, entre autres, un DIN ou un DIN et un AC, étant donné que ces types d'exportation commerciale sont généralement considérés comme des ventes au Canada.
Dans ce cas, la vente du médicament exporté n'est pas considérée comme ayant cessé et le fabricant n'est pas tenu de soumettre un avis de cessation de la vente à Santé Canada. L'état dans la BDPP continuera d'indiquer que ce médicament est Commercialisé et le fabricant se doit continuer à remplir ses obligations après la commercialisation.
6.9.2 Exportation en vertu de l'article 37
Lorsque la vente d'un médicament destiné à la consommation au Canada a cessé, mais que le fabricant continue de l'exporter en vertu de l'article 37 de la Loi sur les aliments et drogues, la vente du médicament est considérée comme ayant cessé au Canada.
Dans ce cas, les fabricants devront envoyer un avis de cessation de la vente à Santé Canada. Ce n'est que lorsqu'un avis de cessation de la vente complet et dûment rempli reçu et traité que l'état dans la BDPP indique que le médicament est Annulé (après commercialisation).
6.10 Soumission des avis à Santé Canada
Pour que Santé Canada puisse traiter les avis reçus du fabricant de façon rapide et efficace, tous les documents requis doivent être fournis, dûment remplis et soumis dans le format approprié.
6.10.1 Documents requis
| Activité | FDM | Étiquette |
|---|---|---|
| Avis de mise en marché | Requis | Consulter le tableau 3 |
| Avis de 12 mois sans vente | Non requis | Non requis |
| Avis de cessation de la vente | Non requis | Non requis |
| Demande de réémission d'un DIN | Non requis | Non requis |
Lorsqu'un fabricant présente un avis ou une demande de réémission de DIN à Santé Canada, celui-ci doit fournir tous les documents et tous les renseignements requis (consultez le tableau 4). Si les documents fournis ne sont pas dûment remplis ou si des renseignements requis sont manquants ou inexacts, l'avis ne pourra pas être traité. Ce n'est lorsque Santé Canada recevra tous les documents et tous les renseignements requis que l'avis pourra être traité.
Une confirmation sera envoyée au fabricant par courriel lorsque le traitement des avis ou des demandes de réémission de DIN ont été complétés, excepté pour les avis de mise en marché. Le fabricant peut confirmer le changement de l'état en consultant la BDPP.
6.10.2 Format et directives relatives à la soumission des avis
Les avis de mise en marché, les avis de cessation de la vente, les avis de 12 mois sans vente et les demandes de réémission d'un DIN doivent tous être envoyés sous format électronique. Des duplicatas ne doivent pas être envoyés en format papier puisque Santé Canada n'accepte plus les copies papier. Comme tout renseignement associé aux présentations de drogue et aux demandes d'autorisation envoyé par voie électronique, les renseignements reçus après 17h00, heure normale de l'Est, le samedi ou le dimanche ou lors des jours fériés seront considérés comme ayant été reçus lors du prochain jour ouvrable.
Les documents concernant les présentations soumises en format eCTD doivent être préparés et soumis conformément à la Ligne directrice : Préparation des activités de réglementation des drogues en format Electronic Common Technical Document.
Les documents concernant les présentations soumises en format électronique autre que le format eCTD doivent être préparés et soumis conformément à la Ligne directrice : Préparation des activités de réglementation en format « Électronique autre que le format eCTD ».
Annexe A Modèle : Demande électronique de réémission d'un DIN
Objet : Demande de réémission d'un DIN pour [nom du produit - DIN XXXXXXXX]
Madame, Monsieur,
Le présent courriel vise à faire une demande de réémission du DIN XXXXXXXX pour [nom du produit].
Les renseignements ci-après sont fournis pour aider Santé Canada à déterminer si la soumission d'une présentation de drogue ou d'une demande d'autorisation est requise pour la réémission du DIN.
- Y a-t-il eu des changements dans la formulation du médicament depuis la cessation de sa vente? O/N
- Y a-t-il eu des changements dans la fabrication du médicament depuis la cessation de sa vente? O/N
- Est-ce que le produit est disponible dans d'autres jurisdictions (p. ex. la Food and Drug Administration des États-Unis)? O/N
- Y a-t-il eu des changements dans l'étiquetage du médicament depuis la cessation de sa vente? O/N
- Y a-t-il eu des réactions indésirables inattendues déclarées à l'échelle nationale ou internationale pour ce médicament? O/N
- Quelles sont les raisons ayant menées à l'annulation de ce DIN?
J'atteste que les renseignements fournis sont justes et exacts.
Cordialement,
[Signature autorisée]